Диметиланилин — Википедия (с комментариями)
Материал из Википедии — свободной энциклопедии
Диметиланилин (N,N-диметиланилин) — органическое соединение, принадлежащее классу третичных аминов. Формально является производным аммиака, в котором атомы водорода замещены на фенильный и два метильных радикала.
Физические свойства
Бесцветная или желтоватая жидкость с неприятным аминным запахом слаборастворимая в воде (1-2%), но растворяется в большинстве органических растворителей, например, метаноле, этиловом спирте, диэтиловом эфире[1], ацетоне и бензоле. При хранении на свету и доступе воздуха темнеет. Кипит при 194°C и атмосферном давлении.
Химические свойства
Слабое основание pKb=5,06 (25°C, вода), слабее диметиламина (pKb=3,23) из-за эффекта сопряжения с фенильной группой, но значительно сильнее анилина (pKb=9,40) из-за индуктивного эффекта двух метильных групп (электронодоноры). Образует соли с сильными минеральными кислотами. Соль с йодоводородной кислотой может быть использована для очистки диметиланилина.
Нитрозируется в пара-положение. Под действием фосгена образует кетон Михлера. Под действием алкилирующих агентов кватернизуется, например под действием метилового эфира p-толуолсульфокислоты
Методы синтеза
Реакцией анилина с метилирующими агентами, например метилиодидом, как и был впервые получен Гофманом (A. W. Hofmann) в 1850-ом году:
- C6H5NH2 + 2 CH3I → C6H5N(CH3)2 + 2 HI
или диметилсульфатом[2]. Также используется реакция анилина с метиловым спиртом в присутствии серной кислоты при нагревании до 210°C и под давлением до 30 атмосфер[3].
- C6H5NH2 + 2 CH3OH → C6H5N(CH3)2 + 2 H2O
Применение
Применяется в производстве полиэфирных смол и в органическом синтезе, особенно в синтезе красителей. Например, малахитового зелёного, метиленового синего. Также используется для производства тетрила (взрывчатое вещество) [4].
Безопасность
Действие на человека аналогично анилину. Попадает в организм как при вдыхании паров, так и через неповреждённую кожу. ЛД50 1410 мг/кг (крысы перорально). Довольно горюч (температура вспышки 63°C).
Напишите отзыв о статье «Диметиланилин»
Примечания
- ↑ Справочник химика. — Т.2. — Л.-М.: Химия, 1964. — С. 426-427
- ↑ 1 2 В. Хиккинботтом. Реакции органических соединений. ГОНТИ. НКТП. 1939. стр. 352
- ↑ К. Вейганд. Методы эксперимента в органической химии. Часть 2. Методы синтеза. М., ИЛ, 1952, стр. 254
- ↑ Химическая энциклопедия. — Т.1. — М.: Советская энциклопедия, 1988. — С. 89
См. также
Отрывок, характеризующий Диметиланилин
– Вообще этого кровожадного солдата, – сказал Билибин, – надо обратить к более человеколюбивым взглядам.– Куда?
– К императору.
– О! о! о!
– Ну, до свидания, Болконский! До свидания, князь; приезжайте же обедать раньше, – пocлшaлиcь голоса. – Мы беремся за вас.
– Старайтесь как можно более расхваливать порядок в доставлении провианта и маршрутов, когда будете говорить с императором, – сказал Билибин, провожая до передней Болконского.
– И желал бы хвалить, но не могу, сколько знаю, – улыбаясь отвечал Болконский.
– Ну, вообще как можно больше говорите. Его страсть – аудиенции; а говорить сам он не любит и не умеет, как увидите.
На выходе император Франц только пристально вгляделся в лицо князя Андрея, стоявшего в назначенном месте между австрийскими офицерами, и кивнул ему своей длинной головой. Но после выхода вчерашний флигель адъютант с учтивостью передал Болконскому желание императора дать ему аудиенцию.
Император Франц принял его, стоя посредине комнаты. Перед тем как начинать разговор, князя Андрея поразило то, что император как будто смешался, не зная, что сказать, и покраснел.
Князь Андрей отвечал. После этого вопроса следовали другие, столь же простые вопросы: «здоров ли Кутузов? как давно выехал он из Кремса?» и т. п. Император говорил с таким выражением, как будто вся цель его состояла только в том, чтобы сделать известное количество вопросов. Ответы же на эти вопросы, как было слишком очевидно, не могли интересовать его.
– В котором часу началось сражение? – спросил император.
– Не могу донести вашему величеству, в котором часу началось сражение с фронта, но в Дюренштейне, где я находился, войско начало атаку в 6 часу вечера, – сказал Болконский, оживляясь и при этом случае предполагая, что ему удастся представить уже готовое в его голове правдивое описание всего того, что он знал и видел.
– Сколько миль?
– Откуда и докуда, ваше величество?
– От Дюренштейна до Кремса?
– Три с половиною мили, ваше величество.
– Французы оставили левый берег?
– Как доносили лазутчики, в ночь на плотах переправились последние.
– Достаточно ли фуража в Кремсе?
– Фураж не был доставлен в том количестве…
Император перебил его.
– В котором часу убит генерал Шмит?…
– В семь часов, кажется.
– В 7 часов. Очень печально! Очень печально!
Император сказал, что он благодарит, и поклонился. Князь Андрей вышел и тотчас же со всех сторон был окружен придворными. Со всех сторон глядели на него ласковые глаза и слышались ласковые слова. Вчерашний флигель адъютант делал ему упреки, зачем он не остановился во дворце, и предлагал ему свой дом. Военный министр подошел, поздравляя его с орденом Марии Терезии З й степени, которым жаловал его император. Камергер императрицы приглашал его к ее величеству. Эрцгерцогиня тоже желала его видеть. Он не знал, кому отвечать, и несколько секунд собирался с мыслями. Русский посланник взял его за плечо, отвел к окну и стал говорить с ним.
wiki-org.ru
Диметиланилин Википедия
Диметиланилин (N,N-диметиланилин) — органическое соединение, принадлежащее классу третичных аминов. Формально является производным аммиака, в котором атомы водорода замещены на фенильный и два метильных радикала.
Физические свойства
Бесцветная или желтоватая жидкость с неприятным аминным запахом слаборастворимая в воде (1-2%), но растворяется в большинстве органических растворителей, например, метаноле, этиловом спирте, диэтиловом эфире[2], ацетоне и бензоле. При хранении на свету и доступе воздуха темнеет. Кипит при 194°C и атмосферном давлении.
Химические свойства
Слабое основание pKb=5,06 (25°C, вода), слабее диметиламина (pKb=3,23) из-за эффекта сопряжения с фенильной группой, но значительно сильнее анилина (pKb=9,40) из-за индуктивного эффекта двух метильных групп (электронодоноры). Образует соли с сильными минеральными кислотами. Соль с йодоводородной кислотой может быть использована для очистки диметиланилина. Нитрозируется в пара-положение. Под действием фосгена образует кетон Михлера. Под действием алкилирующих агентов кватернизуется, например под действием метилового эфира p-толуолсульфокислоты[3].
Методы синтеза
Реакцией анилина с метилирующими агентами, например метилиодидом, как и был впервые получен Гофманом (A. W. Hofmann) в 1850-ом году:
- C6H5NH2 + 2 CH3I → C6H5N(CH3)2 + 2 HI
или диметилсульфатом[3]. Также используется реакция анилина с метиловым спиртом в присутствии серной кислоты при нагревании до 210°C и под давлением до 30 атмосфер [4].
- C6H5NH2 + 2 CH3OH → C6H5N(CH3)2 + 2 H2O
Применение
Применяется в производстве полиэфирных смол и в органическом синтезе, особенно в синтезе красителей. Например, малахитового зелёного, метиленового синего. Также используется для производства тетрила (взрывчатое вещество)[5].
Безопасность
Действие на человека аналогично анилину. Попадает в организм как при вдыхании паров, так и через неповреждённую кожу. ЛД50 1410 мг/кг (крысы перорально). Довольно горюч (температура вспышки 63°C).
Примечания
- ↑ http://www.cdc.gov/niosh/npg/npgd0223.html
- ↑ Справочник химика. — Т.2. — Л.-М.: Химия, 1964. — С. 426-427
- ↑ 1 2 В. Хиккинботтом. Реакции органических соединений. ГОНТИ. НКТП. 1939. стр. 352
- ↑ К. Вейганд. Методы эксперимента в органической химии. Часть 2. Методы синтеза. М., ИЛ, 1952, стр. 254
- ↑ Химическая энциклопедия. — Т.1. — М.: Советская энциклопедия, 1988. — С. 89
См. также
wikiredia.ru
Диметиланилин — Википедия. Что такое Диметиланилин
Материал из Википедии — свободной энциклопедии
Диметиланилин (N,N-диметиланилин) — органическое соединение, принадлежащее классу третичных аминов. Формально является производным аммиака, в котором атомы водорода замещены на фенильный и два метильных радикала.
Физические свойства
Бесцветная или желтоватая жидкость с неприятным аминным запахом слаборастворимая в воде (1-2%), но растворяется в большинстве органических растворителей, например, метаноле, этиловом спирте, диэтиловом эфире[2], ацетоне и бензоле. При хранении на свету и доступе воздуха темнеет. Кипит при 194°C и атмосферном давлении.
Химические свойства
Слабое основание pKb=5,06 (25°C, вода), слабее диметиламина (pKb=3,23) из-за эффекта сопряжения с фенильной группой, но значительно сильнее анилина (pKb=9,40) из-за индуктивного эффекта двух метильных групп (электронодоноры). Образует соли с сильными минеральными кислотами. Соль с йодоводородной кислотой может быть использована для очистки диметиланилина. Нитрозируется в пара-положение. Под действием фосгена образует кетон Михлера. Под действием алкилирующих агентов кватернизуется, например под действием метилового эфира p-толуолсульфокислоты[3].
Методы синтеза
Реакцией анилина с метилирующими агентами, например метилиодидом, как и был впервые получен Гофманом (A. W. Hofmann) в 1850-ом году:
- C6H5NH2 + 2 CH3I → C6H
или диметилсульфатом[3]. Также используется реакция анилина с метиловым спиртом в присутствии серной кислоты при нагревании до 210°C и под давлением до 30 атмосфер[4].
- C6H5NH2 + 2 CH3OH → C6H5N(CH3)2 + 2 H2O
Применение
Применяется в производстве полиэфирных смол и в органическом синтезе, особенно в синтезе красителей. Например, малахитового зелёного, метиленового синего. Также используется для производства тетрила (взрывчатое вещество)[5].
Безопасность
Действие на человека аналогично анилину. Попадает в организм как при вдыхании паров, так и через неповреждённую кожу. ЛД50 1410 мг/кг (крысы перорально). Довольно горюч (температура вспышки 63°C).
Примечания
- ↑ http://www.cdc.gov/niosh/npg/npgd0223.html
- ↑ Справочник химика. — Т.2. — Л.-М.: Химия, 1964. — С. 426-427
- ↑ 1 2 В. Хиккинботтом. Реакции органических соединений. ГОНТИ. НКТП. 1939. стр. 352
- ↑ К. Вейганд. Методы эксперимента в органической химии. Часть 2. Методы синтеза. М., ИЛ, 1952, стр. 254
- ↑ Химическая энциклопедия. — Т.1. — М.: Советская энциклопедия, 1988. — С. 89
См. также
wiki.sc
анилин, N–метиланилин, N,N–диметиланилин, толуидины, фенетидины, дифениламин.
Амины: классификация, номенклатура; химическая идентификация, спектральные характеристики. Представители: анилин, N–метиланилин, N,N–диметиланилин, толуидины, фенетидины, дифениламин.
Аминами называются производные аммиака, в которых один, два иди три атома водорода заменены на углеводородные радикалы.
В зависимости от числа углеводородных радикалов различают первичные, вторичные и третичные амины. Существуют также четвертичные аммониевые соли и основания, представляющие собой производные иона аммония, в котором все четыре атома водорода замещены органическими радикалами. По природе радикала амины подразделяются на алифатические и ароматические.
Для аминов более употребительны названия, построенные по радикально-функциональной, а не по заместительной номенклатуре.
Родовое название амины относится к соединениям RNH2, RR’NH и RR’R»N, которые являются первичными, вторичными и третичными аминами соответственно. В более широком смысле к аминам относятся и соединения, содержащие группу —NH— в цикле.
Названия первичных аминов образуются добавлением суффикса -амин к названию радикала R (способ а) или к названию родоначальной структуры (способ б). Так, соединение CH3CH2CH2NH2 будет называться пропиламин (а) или пропанамин-1 (б). Способ а обычно используют для производных простых соединений, а способ б — для сложных циклических соединений. В способе а применяется принцип замещения атома водорода в молекуле аммиака, который формально является родоначальной структурой. По сути он похож на принцип радикально-функциональной номенклатуры, но в правилах ИЮПАК относится к заместительной.
В тех случаях, когда группа —NH2 не является старшей, она обозначается префиксом амино-:
Некоторые амины сохраняют тривиальные названия:
Первичные диамины и полиамины, в которых все аминогруппы присоединены к алифатической цепи или циклическому ядру, называют путем прибавления суффиксов -диамин, -триамин и т. д. к названию родоначальной структуры или многовалентного радикала. Тривиальное название «бензидин» сохраняется.
Симметричные вторичные и третичные амины называют, присоединяя умножающие приставки ди- или три- к названиям алкильных радикалов с суффиксом -амин. Несимметричные соединения получают названия как Ж-замещенные производные первичных аминов, причем за исходный первичный амин принимают соединение с более сложным радикалом:
Радикалы аминов RNH—, R2N—, RR’N— называют как замещенные аминогруппы или к тривиальным названиям аминов добавляют букву о:
Низшие алифатические амины — газы или жидкости с запахом, похожим на запах аммиака. Высшие гомологи алифатических аминов и ароматические амины представляют собой жидкости или твердые вещества. Амины образуют слабые водородные связи и непрочные ассоциаты, поэтому их температуры кипения ниже, чем у спиртов и карбоновых кислот с тем же числом атомов углерода, но выше, чем у альдегидов или простых эфиров. Низшие алифатические амины хорошо растворимы в воде, с увеличением числа углеводородных радикалов и их длины растворимость снижается. Ароматические амины плохо растворимы в воде.
Представители.
Анилин — C6H5NH2 — бесцветная жидкость со слабым запахом, похожим на запах бензола, при стоянии на воздухе довольно быстро окисляется и приобретает желто-коричневую окраску и неприятный запах. Токсичен.
Более половины производимого анилина расходуется на производство стабилизаторов и ускорителей вулканизации каучуков. Второй по значимости сферой его применения является производство изоцианатов, используемых для получения полиуретанов. Применяют также в производстве красителей различных классов, лекарственных средств, фотоматериалов и средств защиты растений. В нашей стране анилин используют для получения капролактама.
N–метиланилин (монометиланилин) — С6H5NHCH3 — представляет собой маслянистую жидкость желтого цвета с плотностью 0,98 г/см3, растворимую в бензинах, спиртах и эфирах. Главной задачей монометиланилина является получение необходимых детонационных свойств бензина при его производстве. Кроме того, при добавлении его в топливо регулируется октановое число продукта и его экологичность.
Диметиланилин — C6H5N(CH3)2 — третичный жирноароматический амин, бесцветная жидкость. Применяется в производстве полиэфирных смол и в органическом синтезе. Диметиланилин применяют в синтезе красителей (малахитовый зелёный, метиленовый голубой и др.), взрывчатых веществ и др.
Толуидины — CH3C6H4NH2 — бесцветные кристаллические соединения со своеобразными запахами, на воздухе быстро окисляются и темнеют. Получают восстановлением нитротолуолов. Применяют в производстве красителей разных классов (трифенилметановых, азокрасителей, тиазиновых, сернистых), а также для получения крезолов. Толуидины, как и некоторые другие ароматические амины, ядовиты и канцерогенны.
Фенетидины (этоксианилины, аминофенетолы) — NH2–C6H4–OC2H3 (орто-, пара- и мета-) — представляют жидкости. Применяют в производстве азотолов; n-фенетидин также в синтезе лекарственныз средствв (фенацетина, риванола). Фенетидины вызывают отравление при попадании на кожу и вдыхании паров, поражают печень и почки.
Дифенилами́н ((N-фенил)-анилин) — (С6Н5)2NН — бесцветные кристаллы, темнеющие на свету. Дифениламин — исходный продукт в производстве антиоксидантов для полимеров; стабилизатор и флегматизатор термо- и атмосферостойкости нитратов целлюлозы, в том числе пироксилиновых порохов; промежуточный продукт в синтезе триарилметановых и азокрасителей, инсектицидов; ингибитор коррозии мягких сталей. Используется в аналитической химии для обнаружения ионов, как окислительно-восстановительный индикатор.
Нуклеофильные свойства.
Нуклеофильные свойства аминов, как и основные, обусловлены наличием неподеленной пары электронов атома азота. Некоторые реакции, в которых амины участвуют в качестве нуклеофильных реагентов, будут далее и были ранее. Это — алкилирование аминов [(1)], взаимодействие с карбонильными соединениями [(2)] и ацилирование производными карбоновых кислот [(3)]:
Алкилирование. Амины, как и аммиак, подвергаются алкилированию галогеноалканами. Алкилирование аммиака приводит к образованию первичного амина, из первичных аминов образуются вторичные, из вторичных — третичные, из третичных — четвертичные аммониевые соли. Образующаяся в ходе реакции замещенная аммониевая соль обменивается протоном с аммиаком или амином, поэтому в реакции алкилирования получается смесь аминов с разным числом алкильных радикалов:
Реакцию алкилирования редко удается остановить на какой-то отдельной стадии ввиду того, что различия в нуклеофильности и основности первичных, вторичных и третичных аминов не настолько значительны, чтобы повлиять на различие в скоростях реакций алкилирования аминов разной степени замещения.
В промышленности аммиак и низшие амины алкилируют низшими спиртами в газовой фазе при температуре 300—500 °С над оксидами алюминия, кремния, тория, хрома и др. При этом образуются смеси первичных, вторичных и третичных аминов. Этим способом получают в основном метил- и этиламины:
Реакции аминов с эпоксидами. При взаимодействии первичных и вторичных аминов с эпоксидами (оксиранами) происходит нуклеофильное раскрытие напряженного трехчленного α-оксидного цикла и образуются β-аминоспирты. В замещенных эпоксидах нуклеофильная атака происходит, как правило, на наименее замещенный атом углерода оксидного цикла (правило Красуского):
29. Амины: реакции первичных, вторичных и третичных алифатических и ароматических аминов с азотистой кислотой; карбиламинная реакция (изонитрильная проба).
Нуклеофильные свойства.
Карбиламинная (изонитрильная) реакция (качественная реакция). При взаимодействии первичных аминов с хлороформом в спиртовом растворе щелочи образуются изонитрилы (карбиламины):
Первоначально из хлороформа при действии щелочи в результате α-элиминирования образуется дихлорокарбен — электронодефицитная частица с секстетом электронов у атома углерода. Затем происходит нуклеофильное присоединение амина к дихлорокарбену, после в результате последовательного отщепления 2 молекул HCl образуется изонитрил:
Изонитрилы представляют собой жидкости с отвратительным тошнотворным запахом, ядовиты. Они нестойки, разлагаются при действии кислот на исходный первичный амин и муравьиную кислоту. Карбиламинную реакцию применяют в аналитических целях для обнаружения первичных аминов.
Реакции с азотистой кислотой.
Амины разных типов с азотистой кислотой реагируют неодинаково. Некоторые продукты этих реакций, например соли диазония, имеют широкое практическое применение.
Первичные ароматические амины. В результате реакции первичных ароматических аминов с азотистой кислотой при низких температурах в присутствии сильных минеральных кислот образуются соли арилдиазония, а сама реакция называется реакцией диазотирования:
Реакция диазотирования имеет сложный механизм. Показано, что она имеет третий порядок, а скорость ее зависит от концентрации свободного амина (основания), азотистой и сильной минеральной кислот. Нитрозирующей частицей (электрофилом) в этой реакции в зависимости от условий проведения могут быть протонированная форма азотистой кислоты H2O+NO; оксид азота(III) N2O3; нитрозилхлорид NOC1 или нитрозил-катион N=O+, образующиеся из нитрита натрия и минеральной кислоты:
В сильнокислой среде нитрозирующей частицей является нитрозил-катион, который взаимодействует со свободным амином, находящимся в равновесии с аммониевым ионом. Необходимо акцентировать, что электрофильной атаке подвергается именно свободный амин, а не его соль, несмотря на то что концентрация его в сильнокислой среде может быть очень низкой:
Образующийся катион (I) отщепляет протон и превращается в N-нитрозоамин (II), который перегруппировывается в диазогидроксид (III). Диазогидроксид протонируется и отщепляет воду, превращаясь в катион диазония (IV):
Условия диазотирования конкретного амина зависят от двух факторов — основности и растворимости в воде. Амины с относительно высокой основностью и хорошей растворимостью в воде диазотируют в разбавленных растворах в слабокислой среде. В этих условиях в растворе создается достаточно высокая концентрация свободного амина, а в качестве нитрозирующих агентов выступает свободная или протонированная азотистая кислота, а также оксид азота(III). Амины с низкой основностью, например нитроанилины, диазотируют в концентрированной серной кислоте, в этой среде образуется более сильный электрофил — нитрозил-катион.
Ароматические соли диазония — неустойчивые соединения, в сухом виде взрывчаты, большинство из них хорошо растворимы в воде, поэтому их получают в водных растворах и сразу же используют для дальнейших превращений.
Первичные алифатические амины. Эти амины при действии азотистой кислоты в водных растворах подвергаются дезаминированию. Вначале они, как и ароматические амины, образуют соли диазония, однако последние крайне неустойчивы и разлагаются даже в растворах и при низких температурах. Катион алкилдиазония отщепляет молекулу азота и превращается в соответствующий карбокатион. Образовавшийся карбокатион присоединяет различные нуклеофилы, имеющиеся в реакционной смеси (воду, хлорид- или нитрит-ион), отщепляет протон и превращается соответственно в спирт, алкилгалогенид, нитрозоэфир или этиленовый углеводород. Кроме того, карбокатионы могут претерпевать различные перегруппировки. Перегруппировочные карбокатионы в свою очередь также могут отщеплять протон и присоединять различные нуклеофилы. Таким образом, в результате дезаминирования первичных алифатических аминов образуется, как правило, сложная смесь продуктов, что иллюстрируется схемой дезаминирования бутиламина:
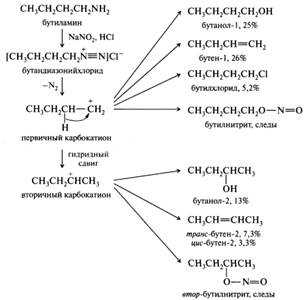
Вторичные амины. Алифатические и ароматические вторичные амины при взаимодействии с азотистой кислотой образуют N-нитрозоамины, представляющие собой нерастворимые в воде маслянистые жидкости или твердые вещества желтого цвета. Эту реакцию можно использовать для идентификации вторичных аминов:
Третичные ароматические амины. Электрофильной атаке нитрозирующим агентом подвергается ароматическое кольцо, при этом образуются пара-нитрозопроизводные:
Третичные алифатические амины. При низких температурах и низкой концентрации азотистой кислоты третичные алифатические амины не реагируют. При нагревании происходит дезалкилирование третичного амина с образованием вторичного N-нитрозоамина и окислением отщепившегося радикала в соответствующий альдегид:
Номенклатура.
Используемый термин «родоначальная молекула» (не путать с родоначальной структурой!) подразумевает молекулы RH и R’H, производными которых будут азосоединения R—N=N—R’.
Названия соединений, в которых азогруппа — N=N— связывает радикалы, производимые от идентичных родоначальных молекул без заместителей, образуются добавлением префикса азо- к названию незамещенной родоначальной молекулы. Заместители обозначаются с помощью префиксов и суффиксов обычным способом. Азогруппа получает наименьшие локанты. Один набор локантов отмечается штрихами:
В тех случаях, когда азогруппа связывает различные радикалы, в названии азосоединения частицу азо- помещают между полными наименованиями родоначальных молекул (замещенных). Если необходимы цифровые указатели для обозначения положения азогруппы, то их помещают между частицей азо- и названиями молекул, к которым эти локанты относятся. Первой указывается более сложная родоначальная молекула. Азогруппа получает наименьшие возможные локанты. Все заместители в первом компоненте обозначаются префиксами (за исключением тех, которые включаются в тривиальное или полутривиальное название этого компонента). Локанты в первом компоненте обозначаются цифрами без штрихов, а во втором — со штрихами:
Другой способ – в названии соединений RN=NR’ радикал RN=N— рассматривают как заместитель в родоначальной молекуле R’H. В качестве R’H выбирают такую молекулу, которая имеет большее число старших характеристических групп. Если их число в обоих компонентах одинаково, то за R’H принимают более сложную молекулу, а группу RN=N— называют R-азо-. Суффикс должен получить наименьший возможный локант; следующие, насколько возможно, низкие номера должна иметь азогруппа:
Если радикалы R и R’ образованы из одной и той же незамещенной родоначальной молекулы и несут одинаковое число характеристических групп, обозначаемых суффиксами, то названию незамещенного азосоединения предшествует префикс азоди-, а перед ним перечисляются префиксы других заместителей. Суффикс, а за ним азогруппа должны иметь по возможности меньшие локанты. Как и в первом способе, используются цифровые указатели со штрихами и без них:
Реакции аминов с азотистой кислотой.
Первичные ароматические амины. В результате реакции первичных ароматических аминов с азотистой кислотой при низких температурах в присутствии сильных минеральных кислот образуются соли арилдиазония, а сама реакция называется реакцией диазотирования:
Реакция диазотирования имеет сложный механизм. Показано, что она имеет третий порядок, а скорость ее зависит от концентрации свободного амина (основания), азотистой и сильной минеральной кислот. Нитрозирующей частицей (электрофилом) в этой реакции в зависимости от условий проведения могут быть протонированная форма азотистой кислоты H2O+NO; оксид азота(III) N2O3; нитрозилхлорид NOC1 или нитрозил-катион N=O+, образующиеся из нитрита натрия и минеральной кислоты:
В сильнокислой среде нитрозирующей частицей является нитрозил-катион, который взаимодействует со свободным амином, находящимся в равновесии с аммониевым ионом. Необходимо акцентировать, что электрофильной атаке подвергается именно свободный амин, а не его соль, несмотря на то что концентрация его в сильнокислой среде может быть очень низкой:
Образующийся катион (I) отщепляет протон и превращается в N-нитрозоамин (II), который перегруппировывается в диазогидроксид (III). Диазогидроксид протонируется и отщепляет воду, превращаясь в катион диазония (IV):
Условия диазотирования конкретного амина зависят от двух факторов — основности и растворимости в воде. Амины с относительно высокой основностью и хорошей растворимостью в воде диазотируют в разбавленных растворах в слабокислой среде. В этих условиях в растворе создается достаточно высокая концентрация свободного амина, а в качестве нитрозирующих агентов выступает свободная или протонированная азотистая кислота, а также оксид азота(III). Амины с низкой основностью, например нитроанилины, диазотируют в концентрированной серной кислоте, в этой среде образуется более сильный электрофил — нитрозил-катион.
Ароматические соли диазония — неустойчивые соединения, в сухом виде взрывчаты, большинство из них хорошо растворимы в воде, поэтому их получают в водных растворах и сразу же используют для дальнейших превращений.
Ароматические диазосоединения.
Более значимыми в практическом отношении являются диазосоединения ароматического ряда. Общая формула ароматических диазосоединений ArN2X, где X — анион сильной кислоты или ковалентносвязанная группа, например, гидроксильная. Строение диазосоединения существенно зависит от характера частицы X. В тех случаях, когда она представляет собой анион сильной кислоты (HSО4—, Сl—, СlO4—), диазосоединение существует в виде ионно построенной соли диазония.
Атомы азота в катионе диазония линейно расположены в плоскости бензольного кольца. Расстояние между атомами азота приблизительно равно 0,109 нм, т. е. по характеру эта связь приближается к тройной. Положительный заряд в катионе распределен в основном на обоих атомах азота, но частично он компенсируется и за счет π-электронного облака ароматического кольца, и таким образом, можно представить следующие резонансные структуры катиона бензолдиазония:
Поскольку первые две структуры вносят наибольший вклад в резонансный гибрид, то катион арилдиазония часто записывается как ArN2+.
Соли диазония устойчивы только при низких температурах (0—5 °С). В сухом виде взрываются даже при низких температурах, поэтому их растворы, как правило водные, готовят по мере надобности и долго не хранят. Комплексы солей диазония с некоторыми кислотами Льюиса — двойные диазониевые соли — сравнительно устойчивы и могут довольно долго сохраняться в сухом виде при комнатной температуре. К ним относятся соли с такими анионами, как BF4—, ZnCl3—, SbCl4—, SbCl6—, HgCl3—, FeCl4—, например:
Соли диазония, получаемые из ариламиносульфоновых кислот, существуют в виде диполярных ионов; вероятно, поэтому многие из них малорастворимы в воде.
Строение диазосоединений зависит от pH среды: в кислой среде они существуют в виде солей диазония, при подщелачивании раствора соли диазония превращаются в ковалентно построенные диазогидроксиды. Диазогидроксиды обладают кислотными свойствами, поэтому при дальнейшем добавлении щелочи они отщепляют протон и образуют соли — диазотаты. При подкислении растворов диазотатов снова образуются диазогидроксиды и соли диазония:
Индикаторные свойства.
Еще более ста лет назад окраску веществ связывали с наличием в их структуре так называемых хромофорных групп, к которым относятся некоторые ненасыщенные группировки, например, двойные связи С=С, С=O, C=N, N=N, N=O, ароматические фрагменты.
Изолированные хромофоры имеют полосы поглощения в электронном спектре в дальней ультрафиолетовой области (165—200 нм) и являются прозрачными в видимой области спектра. Сопряжение одного хромофора с другим вызывает сдвиг полос поглощения в сторону больших длин волн с одновременным увеличением их интенсивности. Окрашенные вещества поглощают в видимой области спектра (400—800 нм). Очевидно, что такие соединения должны иметь в своей структуре длинную цепь сопряжения. Типичным примером окрашенных веществ служат азосоединения, характеризующиеся наличием в структуре в качестве главного хромофора фрагментаазобензола. Сопряженная система азобензола включает два бензольных кольца и азогруппу:
Различные азосоединения в зависимости от длины сопряженной системы могут быть окрашены в желтый, оранжевый, красный, синий и зеленый цвета. Изменению и углублению окраски способствует наличие в структуре ауксохромов — атомов или групп атомов, вступающих в р,π- и π,π-сопряжение с π-электронной системой главного хромофора. Наиболее интенсивную окраску имеют те соединения, в которых с главным хромофором сопряжены одновременно электронодонорные и электроноакцепторные группы, находящиеся в пара- или орто-положении по отношению друг к другу. Примерами таких соединений могут служить метиловый оранжевый (гелиантин), имеющий электронодонорную диметиламиногруппу, и электроноакцепторную сульфогруппу, а также метиловый красный, конго красный и др.
Многие ароматические азосоединения при действии кислот и оснований изменяют свою окраску в определенном интервале pH среды, благодаря чему используются в аналитической химии как индикаторы. При изменении pH происходит протонирование или депротонирование молекулы индикатора, что влечет за собой перераспределение электронной плотности в сопряженной системе.
Например, индикатор метиловый оранжевыйв нейтральной и щелочной средах окрашен в желтый цвет. В кислой среде вследствие протонирования одного из атомов азота происходит изменение в распределении электронной плотности в сопряженной системе молекулы, являющееся причиной изменения желтой окраски на красную.
Протонированная форма молекулы метилового оранжевого может быть описана двумя мезомерными структурами, одна из которых хиноидная. Полагают, что изменение окраски в основном обусловлено вкладом хиноидной структуры (выделена цветом):
Имеется большое число кислотно-основных индикаторов, относящихся к разным классам окрашенных веществ, интервалы перехода окраски которых перекрывают весь диапазон значений pH. Большую ценность представляют индикаторы с узким интервалом перехода, например нитразиновый желтый.
Альдегиды и кетоны: классификация; номенклатура; химическая идентификация, спектральные характеристики. Представители: формальдегид (формалин), ацетальдегид, хлораль (хлоральгидрат), акролеин, бензальдегид, ацетон, циклогексанон, ацетофенон.
Альдегидами называются соединения, в которых карбонильная группа соединена с углеводородным ради капом и атомом водорода, а кетонами карбонильные соединения с двумя углеводородными радикалами. В альдегидах и кетонах функциональной группой является карбонильная группа >С=O, поэтому оба класса этих родственных веществ относятся к карбонильным соединениям:
В зависимости от строения углеводородных радикалов альдегиды и кетоны бывают алифатическими (насыщенными и ненасыщенными) и ароматическими. Кетоны, у которых карбонильная группа соединена с одинаковыми углеводородными радикалами, называются симметричными.
Номенклатура. Родовое название альдегиды имеют соединения, у которых группа — СН=О присоединена к атому углерода. В названиях ациклических альдегидов группа —СНО, если она является старшей и находится в главной цепи, обозначается суффиксом -аль. Нумерация ведется в этом случае от нее, поэтому локант «1» опускается. Диальдегиды называют путем добавления суффикса -диаль к названию родоначальной структуры. Если группа —СНО не является старшей или находится не в главной цепи, то используют префикс формил-.
Названия циклических альдегидов, в которых группа —СНО как старшая связана с циклом, строятся добавлением суффикса -карбальдегид к названию циклической системы:
Если соответствующая альдегиду карбоновая кислота имеет тривиальное название, то из него может быть образовано и тривиальное название альдегида:
Сохраняются следующие тривиальные названия:
Родовое название кетоны имеют соединения, содержащие карбонильную группу >С=О, связанную с двумя атомами углерода (оксогруппой называется фрагмент =O). Кетоны называют, используя суффикс -он, если нет более старшей группы. В ее присутствии используется префикс оксо-.
Сохраняется тривиальное название «ацетон» для СН3СОСН3.
Кетоны Ar— СО—R, в которых карбонильная группа присоединена к бензольному или нафталиновому ядру, называют, заменяя частицу -ил в названии ацильного радикала R—СО— на суффиксы -офенон и -онафтон соответственно:
Дикетоны, производимые от ароматических соединений заменой двух фрагментов — СН= на группы >С=O с последующей перегруппировкой двойных связей, называют, добавляя суффикс -хинон к названию ароматического соединения (иногда это название подвергают модификации):
Низшие алифатические альдегиды и кетоны, за исключением газообразного формальдегида, представляют собой подвижные жидкости. Первые представители (формальдегид, ацетальдегид, ацетон) хорошо растворимы в воде за счет образования с ней водородных связей или гидратных форм. По мере удлинения углеродной цепи растворимость карбонильных соединений в воде снижается. Ароматические карбонильные соединения плохо растворимы в воде.

Отдельные представители.
Формальдегид — СН2О — простейший и единственный газообразный альдегид, обладает резким раздражающим запахом, хорошо растворим в воде и спиртах, хуже в эфире, бензоле. В промышленности формальдегид получают парофазным окислением метанола, железо-молибденовые катализаторы обеспечивают выходы до 92%:
Формальдегид склонен к полимеризации, в зависимости от условий образуется линейный олигомер (параформальдегид, или параформ) или циклические тример и тетрамер. Параформ представляет собой белый порошок, при нагревании разлагается с образованием формальдегида, поэтому используется как форма хранения и транспортировки формальдегида.
При взаимодействии формальдегида с аммиаком образуется гексаметилентетрамин (уротропин). Первоначально образующийся продукт нуклеофильного присоединения аммиака к альдегиду в несколько стадий превращается в уротропин, каркасная структура которого сходна с кристаллической решеткой алмаза:
Гексаметилентетрамин используется в медицине как антисептическое средство. Основная масса производимого промышленностью формальдегида используется для производства феноло- и мочевиноформальдегидных смол. Водный 40%-й раствор формальдегида, стабилизированный добавкой 6—10% метанола, называется формалином (антисептик).
Ацетальдегид — СН3СНО — низкокипящая жидкость с резким запахом, смешивается во всех отношениях с водой и большинством органических растворителей. Ацетальдегид, подобно формальдегиду, в присутствии кислот легко образует циклический тример (паральдегид) и тетрамер (метальдегид):
Метальдегид используется в качестве топлива (сухой спирт).
Ацетальдегид служит сырьем для промышленного получения многих соединений алифатического ряда — уксусной кислоты, уксусного ангидрида, этилацетата, бутанола-1, хлораля.
Хлораль (трихлорацетальдегид, трихлоруксусный альдегид) — CCl3CHO — бесцветная жидкость со специфическим резким запахом, растворим в органических растворителях и нерастворим в воде. Хлораль используют в производстве инсектицидов, в частности в производстве ДДТ, хлорофоса, дихлофоса.
Хлоральгидрат (2,2,2-трихлорэтандиол-1,1, C(Сl)3С(ОН)2Н) обладает снотворным и седативным действием, использующихся при анестезии. Чаще хлоралгидрат используется при психическом возбуждении и как противосудорожное средство при спазмофилии,столбняке и т. п. Входит в состав зубных капель «Дента».
Бензальдегид — С6Н5СНО — бесцветная жидкость с запахом горького миндаля. Пары бензальдегида обладают слезоточивым действием. Растворим в этаноле, диэтиловом эфире и других органических растворителях, практически нерастворим в воде. В природе встречается в виде гидроксинитрила C6H5CH(OH)CN (как составная часть амигдалина). Бензальдегид используют для синтеза коричной кислоты и альдегида, бензилбензоата, трифенилметановых красителей.
Акролеин (акриловый альдегид) — СН2=СНСНО — простейший ненасыщенный альдегид, представляющий собой бесцветную слезоточивую жидкость с резким запахом, образуется при термическом разложении жиров (кухонный чад), растворим в воде и органических растворителях. Акриловый альдегид легко полимеризуется и окисляется, поэтому его хранят в присутствии ингибиторов радикальных реакций. Применяют акриловый альдегид для синтеза акрилонитрила, глицерина, пиридина, некоторых аминокислот.
Ацетон — СН3СОСН3 — простейший кетон, бесцветная жидкость с характерным запахом, смешивается с водой и органическими растворителями. Образуется как продукт аномального расщепления углеводов у больных сахарным диабетом (ацетоновые тела). Ацетон находит широкое применение как растворитель лаков и красок, служит сырьем для синтеза уксусного ангидрида и кетена СН2=С=О.
Ацетофенон (метилфенилкетон) — C6H5COCH3 — бесцветная маслянистая жидкость, обладающая сильным запахом черёмухи. Хорошо растворяется в этаноле, диэтиловом эфире, ацетоне, хлороформе, бензоле. Ацетофенон и некоторые его производные используются как душистые вещества в парфюмерии. Кроме того, ацетофенон обладает снотворным действием. Его производное — хлорацетофенон — является слезоточивым веществом.
Циклогексанон — жидкость с раздражающим запахом (ацетон и мята), растворим в органических растворителях, ограниченно растворим в воде. Получают гидрированием фенола с последующим окислением или дегидрированием циклогексанола. Применяют для получения ɛ-капролактама и адипиновой кислоты, например:
Спектральная идентификация
ИК-спектроскопия. В ИК-спектрах альдегидов и кетонов имеется сильная полоса валентных колебаний группы С=О. У алифатических альдегидов максимум полосы поглощения находится около 1725 см-1, у кетонов 1715 см-1. Если карбонильная группа сопряжена с С = С или ароматической системой, максимум полосы поглощения смещается в низкочастотную область: у ароматических альдегидов — 1715-1695 см-1, у α,β-ненасышенных альдегидов — 1710-1685 см-1, у алкил-арилкетонов — около 1690 см-1, у диарилкетонов — около 1665 см-1, у α,β-ненасыщенных кетонов — ~1665 см-1. Для карбонильных групп хинонов характерна полоса поглощения при 1690—1660 см-1. Две полосы слабой интенсивности в области 2720-2690 и 2830-2810 см-1 соответствует валентным колебаниям связи С—Н альдегидов.
Спектроскопия ПМР. В спектрах ПМР альдегидов наиболее характеристичными являются сигналы протона альдегидной группы в интервале 9,4-10,4 м. д. (9,72 м. д. у ацетальдегида, 9,48 м. д. у акрилового альдегида, 9,96 м. д. у бензальдегида). Нахождение этого сигнала в столь слабом поле обусловлено магнитной анизотропией карбонильной группы. Внешнее магнитное поле Н0 индуцирует в π-электронной системе связи С=О циркуляцию электронов, которая в свою очередь создает области, где протоны подвергаются соответственно экранированию и дезэкранированию.
Карбонильная группа за счет индуктивного эффекта дезэкранирует протоны у α-атомов углерода альдегидов и кетонов. Сигналы метальных протонов групп, непосредственно связанных с карбонильной группой, наблюдаются в интервале от 1,9 до 2,2 м, д. (2,07 м. д. у ацетона, 1,93 м. д. у ацетальдегида). Метиленовые группы дают сигнал в более слабом поле, например, в спектре пропионового альдегида протоны метиленовой группы дают сигнал 2,40 м. д. В спектрах ПМР хинонов протоны кольца дают сигналы в области 6,7 м. д.
infopedia.su
Диметиланилин Вики
Диметиланилин (N,N-диметиланилин) — органическое соединение, принадлежащее классу третичных аминов. Формально является производным аммиака, в котором атомы водорода замещены на фенильный и два метильных радикала.
Физические свойства[ | код]
Бесцветная или желтоватая жидкость с неприятным аминным запахом слаборастворимая в воде (1-2%), но растворяется в большинстве органических растворителей, например, метаноле, этиловом спирте, диэтиловом эфире[2], ацетоне и бензоле. При хранении на свету и доступе воздуха темнеет. Кипит при 194°C и атмосферном давлении.
Химические свойства[ | код]
Слабое основание pKb=5,06 (25°C, вода), слабее диметиламина (pKb=3,23) из-за эффекта сопряжения с фенильной группой, но значительно сильнее анилина (pKb=9,40) из-за индуктивного эффекта двух метильных групп (электронодоноры). Образует соли с сильными минеральными кислотами. Соль с йодоводородной кислотой может быть использована для очистки диметиланилина. Нитрозируется в пара-положение. Под действием фосгена образует кетон Михлера. Под действием алкилирующих агентов кватернизуется, например под действием метилового эфира p-толуолсульфокислоты[3].
Методы синтеза[ | код]
Реакцией анилина с метилирующими агентами, например метилиодидом, как и был впервые получен Гофманом (A. W. Hofmann) в 1850-ом году:
- C6H5NH2 + 2 CH3I → C6H5N(CH3)2 + 2 HI
или диметилсульфатом[3]. Также используется реакция анилина с метиловым спиртом в присутствии серной кислоты при нагревании до 210°C и под давлением до 30 атмосфер[4].
- C6H5NH2 + 2 CH3OH → C6H5N(CH3)2 + 2 H2O
Применение[ | код]
Применяется в производстве полиэфирных смол и в органическом синтезе, особенно в синтезе красителей. Например, малахитового зелёного, метиленового синего. Также используется для производства тетрила (взрывчатое вещество)[5].
Безопасность[ | код]
Действие на человека аналогично анилину. Попадает в организм как при вдыхании паров, так и через неповреждённую кожу. ЛД50 1410 мг/кг (крысы перорально). Довольно горюч (температура вспышки 63°C).
Примечания[ | код]
- ↑ http://www.cdc.gov/niosh/npg/npgd0223.html
- ↑ Справочник химика. — Т.2. — Л.-М.: Химия, 1964. — С. 426-427
- ↑ 1 2 В. Хиккинботтом. Реакции органических соединений. ГОНТИ. НКТП. 1939. стр. 352
- ↑ К. Вейганд. Методы эксперимента в органической химии. Часть 2. Методы синтеза. М., ИЛ, 1952, стр. 254
- ↑ Химическая энциклопедия. — Т.1. — М.: Советская энциклопедия, 1988. — С. 89
См. также[ | код]
ru.wikibedia.ru
Диметиланилин — Gpedia, Your Encyclopedia
Текущая версия страницы пока не проверялась опытными участниками и может значительно отличаться от версии, проверенной 17 июня 2015; проверки требуют 2 правки. Текущая версия страницы пока не проверялась опытными участниками и может значительно отличаться от версии, проверенной 17 июня 2015; проверки требуют 2 правки.Диметиланилин (N,N-диметиланилин) — органическое соединение, принадлежащее классу третичных аминов. Формально является производным аммиака, в котором атомы водорода замещены на фенильный и два метильных радикала.
Физические свойства
Бесцветная или желтоватая жидкость с неприятным аминным запахом слаборастворимая в воде (1-2%), но растворяется в большинстве органических растворителей, например, метаноле, этиловом спирте, диэтиловом эфире[2], ацетоне и бензоле. При хранении на свету и доступе воздуха темнеет. Кипит при 194°C и атмосферном давлении.
Химические свойства
Слабое основание pKb=5,06 (25°C, вода), слабее диметиламина (pKb=3,23) из-за эффекта сопряжения с фенильной группой, но значительно сильнее анилина (pKb=9,40) из-за индуктивного эффекта двух метильных групп (электронодоноры). Образует соли с сильными минеральными кислотами. Соль с йодоводородной кислотой может быть использована для очистки диметиланилина. Нитрозируется в пара-положение. Под действием фосгена образует кетон Михлера. Под действием алкилирующих агентов кватернизуется, например под действием метилового эфира p-толуолсульфокислоты[3].
Методы синтеза
Реакцией анилина с метилирующими агентами, например метилиодидом, как и был впервые получен Гофманом (A. W. Hofmann) в 1850-ом году:
- C6H5NH2 + 2 CH3I → C6H5N(CH3)2 + 2 HI
или диметилсульфатом[3]. Также используется реакция анилина с метиловым спиртом в присутствии серной кислоты при нагревании до 210°C и под давлением до 30 атмосфер[4].
- C6H5NH2 + 2 CH3OH → C6H5N(CH3)2 + 2 H2O
Применение
Применяется в производстве полиэфирных смол и в органическом синтезе, особенно в синтезе красителей. Например, малахитового зелёного, метиленового синего. Также используется для производства тетрила (взрывчатое вещество)[5].
Безопасность
Действие на человека аналогично анилину. Попадает в организм как при вдыхании паров, так и через неповреждённую кожу. ЛД50 1410 мг/кг (крысы перорально). Довольно горюч (температура вспышки 63°C).
Примечания
- ↑ http://www.cdc.gov/niosh/npg/npgd0223.html
- ↑ Справочник химика. — Т.2. — Л.-М.: Химия, 1964. — С. 426-427
- ↑ 1 2 В. Хиккинботтом. Реакции органических соединений. ГОНТИ. НКТП. 1939. стр. 352
- ↑ К. Вейганд. Методы эксперимента в органической химии. Часть 2. Методы синтеза. М., ИЛ, 1952, стр. 254
- ↑ Химическая энциклопедия. — Т.1. — М.: Советская энциклопедия, 1988. — С. 89
См. также
www.gpedia.com
Диметиланилин Википедия
| Диметиланилин | |
|---|---|
 | |
| Общие | |
| Систематическое наименование | N,N-диметиланилин |
| Хим. формула | C8H11N |
| Физические свойства | |
| Молярная масса | 121.18 г/моль |
| Плотность | 0.956 г/см³ |
| Энергия ионизации | 7,14 ± 0,01 эВ[1] |
| Термические свойства | |
| Т. плав. | 2 °C |
| Т. кип. | 194 °C |
| Т. всп. | 63 °C |
| Давление пара | 1 мм.рт.ст.(20°C) |
| Химические свойства | |
| Диэлектр. прониц. | 4.82 |
| Оптические свойства | |
| Показатель преломления | 1.55819 |
| Структура | |
| Дипольный момент | 1.577 Д |
| Классификация | |
| Рег. номер CAS | 121-69-7 |
| PubChem | 949 |
| Рег. номер EINECS | 204-493-5 |
| SMILES | CN(C)c1ccccc1 |
| InChI | 1S/C8h21N/c1-9(2)8-6-4-3-5-7-8/h4-7H,1-2h4JLTDJTHDQAWBAV-UHFFFAOYSA-N |
| RTECS | BX4725000 |
| ChEBI | 16269 |
| ChemSpider | 924 |
| Безопасность | |
| ЛД50 | 1410 мг/кг |
| Приводятся данные для стандартных условий (25 °C, 100 кПа), если не указано иного. | |
Диметиланилин (N
ru-wiki.ru