Производство этилового спирта из непищевого сырья
Производство спирта из картофеля, зерна, мелассы, сахарной свеклы требует расхода больших количеств этих ценных видов сырья. Замена такого сырья более дешевым является одним из источников экономии пищевых продуктов и снижения себестоимости спирта. Поэтому в последнее время значительно увеличилось производство технического этилового спирта из непищевого сырья: древесины, сульфитных щелоков и синтетическим путем из этиленсодержащих газов.
Производство спирта из древесины
Гидролизная промышленность выпускает из растительных отходов, содержащих целлюлозу, в частности из древесных отходов, ряд продуктов: этиловый спирт, кормовые дрожжи, глюкозу и др.
На гидролизных заводах целлюлозу гидролизуют минеральными кислотами до глюкозы, которая используется для сбраживания в спирт, выращивания дрожжей и выпуска в кристаллическом виде. Существуют гидролизные заводы различного профиля: гидролизно-спиртовые, гидролизно-дрожжевые, гидролизно-глюкозные.
Гидролизная промышленность комплексно перерабатывает древесину, в результате чего на гидролизно-спиртовых заводах получают, кроме этилового спирта, и другие ценные продукты: фурфурол, лигнин, жидкую углекислоту, кормовые дрожжи.
Сырье гидролизного производства
Сырьем гидролизного производства служит древесина в виде различных отходов лесной и деревообрабатывающей промышленности: опилки, щепа, стружка и др. Влажность древесины колеблется от 40 до 60%. Опилки, перерабатываемые гидролизными заводами, обычно имеют влажность 40- 48%. В состав сухих веществ древесины входят целлюлоза, гемицеллюлозы, лигнин и органические кислоты.
Гемицеллюлозы древесины состоят из гексозанов: маннана, галактане и пентозанов: ксилана, арабана и их метилированных производных. Лигнин представляет собой сложное вещество ароматического ряда, химический состав и строение его еще не установлены.
Лигнин представляет собой сложное вещество ароматического ряда, химический состав и строение его еще не установлены.
Химический состав абсолютно сухой древесины приведен в таблице 1.
Таблица 1 – Химический состав абсолютно сухой древесиныКроме древесины, в качестве сырья для гидролизной промышленности применяются и растительные отходы сельского хозяйства: подсолнечная лузга, кукурузная кочерыжка, хлопковая шелуха, солома зерновых злаков.
Химический состав растительных отходов сельского хозяйства представлен в таблице 2.
Таблица 2 – Химический состав растительных отходов сельского хозяйстваТехнологическая схема комплексной переработки древесины
Технологическая схема комплексной переработки древесины состоит из следующих стадий: гидролиз древесины, нейтрализация и очистка гидролизата; сбраживание гидролизного сусла, перегонка гидролизной бражки.
Измельченную древесину подвергают гидролизу разбавленной серной кислотой при нагревании под давлением. При гидролизе гемицеллюлозы и целлюлоза разлагаются. Гемицеллюлозы превращаются в гексозы: глюкозу, галактозу, маннозу и пентозы: ксилозу и арабинозу; целлюлоза – в глюкозу. Лигнин при гидролизе остается в виде нерастворимого остатка.
При гидролизе гемицеллюлозы и целлюлоза разлагаются. Гемицеллюлозы превращаются в гексозы: глюкозу, галактозу, маннозу и пентозы: ксилозу и арабинозу; целлюлоза – в глюкозу. Лигнин при гидролизе остается в виде нерастворимого остатка.
Гидролиз древесины осуществляют в гидролизном аппарате – стальном цилиндрическом сосуде. В результате гидролиза получают гидролизат, содержащий около 2-3% сбраживаемых моносахаридов и нерастворимый остаток-лигнин. Последний можно использовать непосредственно в производстве строительных плит, в кирпичном производстве, при помоле цемента, в качестве топлива; после соответствующей обработки лигнин может применяться в производстве пластмасс, резиновой промышленности и др.
Полученный гидролизат направляют в испаритель, где пар отделяется от жидкости. Выделяющийся пар конденсируют и используют для выделения из него фурфурола, скипидара и метилового спирта. Затем гидролизат охлаждают до 75-80°С, нейтрализуют в нейтрализаторе известковым молоком до pH 4-4,3 и добавляют питательные соли для дрожжей (сернокислый аммоний, суперфосфат). Полученный нейтрализат отстаивают для освобождения от выпавшего осадка сернокислого кальция и других взвешенных частиц. Осевший осадок сернокислого кальция отделяют, сушат, обжигают и получают алебастр, используемый в строительной технике. Нейтрализат охлаждают до 30-32°С и направляют на брожение. Подготовленный таким образом к брожению гидролизат называется суслом. Брожение гидролизного сусла производят непрерывным способом в бродильных чанах. При этом дрожжи непрерывно циркулируют в системе; дрожжи отделяют от бражки на сепараторах. Выделяющийся при брожении углекислый газ используют для выпуска жидкой или твердой углекислоты. Зрелую бражку, содержащую 1,0-1,5% спирта, направляют для перегонки и ректификации на брагоректификационный аппарат и получают этиловый спирт, метиловый спирт и сивушное масло. Барда, полученная после перегонки, содержит пентозы и ее используют для выращивания кормовых дрожжей.
Полученный нейтрализат отстаивают для освобождения от выпавшего осадка сернокислого кальция и других взвешенных частиц. Осевший осадок сернокислого кальция отделяют, сушат, обжигают и получают алебастр, используемый в строительной технике. Нейтрализат охлаждают до 30-32°С и направляют на брожение. Подготовленный таким образом к брожению гидролизат называется суслом. Брожение гидролизного сусла производят непрерывным способом в бродильных чанах. При этом дрожжи непрерывно циркулируют в системе; дрожжи отделяют от бражки на сепараторах. Выделяющийся при брожении углекислый газ используют для выпуска жидкой или твердой углекислоты. Зрелую бражку, содержащую 1,0-1,5% спирта, направляют для перегонки и ректификации на брагоректификационный аппарат и получают этиловый спирт, метиловый спирт и сивушное масло. Барда, полученная после перегонки, содержит пентозы и ее используют для выращивания кормовых дрожжей.
При переработке по указанной схеме из 1 т абсолютно сухой хвойной древесины можно получить следующие количества товарных продуктов:
- Спирта этилового, л ………………….
 . 187
. 187 - Жидкой углекислоты, кг …………….. 70
- или твердой углекислоты, кг ……… 40
- Дрожжей кормовых, кг…………….. .. 40
- Фурфурола, кг …………………………….9,4
- Скипидара, кг ……………………………0,8
- Термоизоляционных и строительных лигно-плит, м2 …. 75
- Алебастра строительного, кг ……..225
- Сивушного масла, к г ………………..0,3
Производство спирта из сульфитных щелоков
При производстве целлюлозы из древесины по сульфитному способу в качестве отхода получают сульфитный щелок – коричневую жидкость с запахом сернистого газа. Химический состав сульфитного щелока (%): вода – 90, сухие вещества – 10, в том числе производные лигнина – лигносульфонаты – 6, гексозы – 2, пентозы -1 , летучие кислоты, фурфурол и другие вещества – около 1. Длительное время сульфитные щелока спускали в реки, они загрязняли воду и уничтожали рыбу в водоемах. В настоящее время у нас имеется ряд заводов по комплексной переработке сульфитного щелока на этиловый спирт, кормовые дрожжи и сульфитно-бардяные концентраты. Производство спирта из сульфитных щелоков состоит из следующих стадий: подготовка сульфитного щелока к брожению, сбраживание сульфитнощелокового сусла, перегонка зрелой сульфитной бражки.
Производство спирта из сульфитных щелоков состоит из следующих стадий: подготовка сульфитного щелока к брожению, сбраживание сульфитнощелокового сусла, перегонка зрелой сульфитной бражки.
Подготовку сульфитного щелока к сбраживанию осуществляют по непрерывной схеме. Щелок продувают воздухом для удаления летучих кислот и фурфурола, задерживающих процесс брожения. Продутый щелок нейтрализуют известковым молоком и затем выдерживают для укрупнения выпавших кристаллов сернокислого и сернистокислого кальция; при этом добавляют питательные соли для дрожжей (сернокислый аммоний и суперфосфат). Затем щелок отстаивают. Осевший осадок- шлам – спускают в канализацию, а осветленный щелок охлаждают до 30-32°С. Подготовленный таким образом щелок называется суслом. Сусло направляют в бродильное отделение и сбраживают так же, как гидролизаты древесины, или применяют метод с подвижной насадкой. Подвижной насадкой называются волокна целлюлозы, остающиеся в щелоке. Метод брожения с подвижной насадкой основан на свойстве некоторых рас дрожжей сорбироваться на поверхности целлюлозных волокон и образовывать хлопья волокнисто-дрожжевой массы, которая в зрелой бражке быстро и полно оседает на дно чана.

Из сульфитно-бардяных концентратов можно получить ценное ароматическое вещество – ванилин.
Технологическая схема комплексной переработки сульфитных щелоков на этиловый спирт, кормовые дрожжи и сульфитно-бардяные концентраты показана на рисунке 2.
Рисунок 2 – Технологическая схема переработки сульфитных щелоков на спиртПри переработке сульфитных щелоков получают в пересчете на 1т еловой древесины:
- Спирта этилового, л ……………….. 30-50
- Спирта метилового, л …………………… 1
- Жидкой углекислоты, л ………….. 19-25
- Сухих кормовых дрожжей, кг …. 15
- Сульфитно-бардяных концентратов влажностью 20%, кг …. 475
Производство спирта синтетическим методом
Сырьем для производства синтетического этилового спирта служат газы нефтеперерабатывающих заводов, которые содержат этилен. Кроме того, можно использовать и другие этиленсодержащие газы: коксовый газ, получаемый при коксовании угля, и попутные нефтяные газы.
В настоящее время синтетический этиловый спирт получают двумя способами: сернокислотной гидратацией и прямой гидратацией этилена.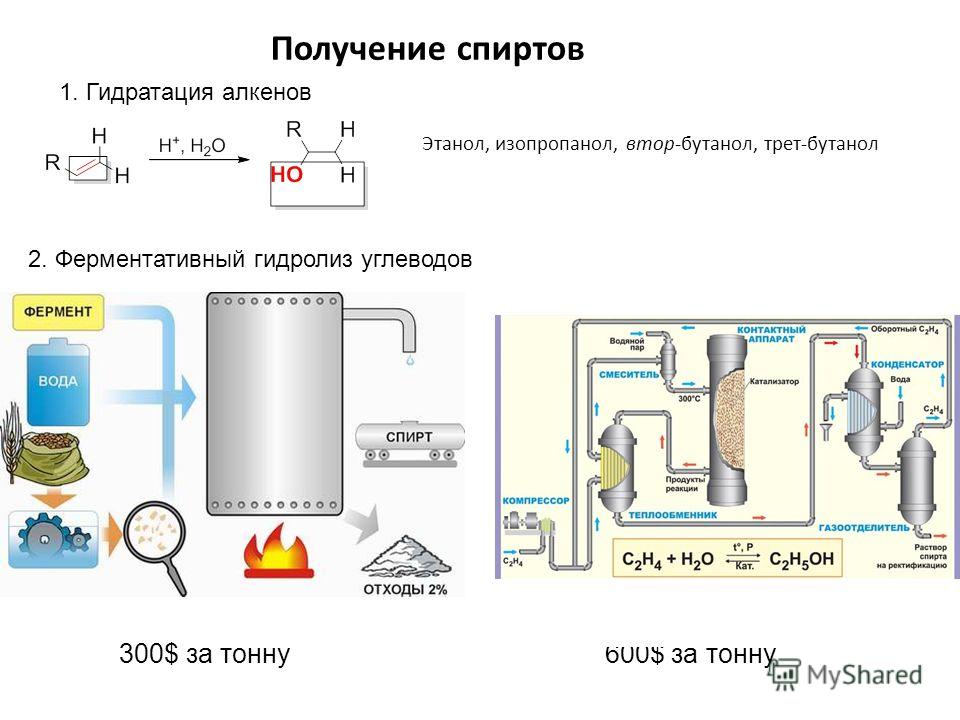
Сернокислая гидратация этилена
Производство этилового спирта этим способом состоит из следующих процессов: взаимодействия этилена с серной кислотой, при котором образуются этилсерная кислота и диэтилсульфат; гидролиз полученных продуктов с образованием спирта; отделение спирта от серной кислоты и очистка его.
Сырьем для сернокислой гидратации служат газы, содержащие 47-50% вес. этилена, а также газы с меньшим содержанием этилена. Процесс осуществляется по схеме, приведенной ниже.
Этилен взаимодействует с серной кислотой в реакционной колонне, представляющей собой вертикальный цилиндр. Внутри колонны находятся колпачковые тарелки с переливными стаканами. В нижнюю часть колонны компрессором подают этиленосодержащий газ, сверху в колонну подводят для орошения 97-98%-ная серная кислота. Газ, поднимаясь вверх, на каждой тарелке барботирует через слой жидкости. Этилен с серной кислотой взаимодействует по реакциям:
Из реакционной колонны непрерывно вытекает смесь этилсерной кислоты, диэтилсульфата и непрореагировавшей серной кислоты. Эту смесь охлаждают в холодильнике до 50°С и направляют на гидролиз, при котором протекают такие реакции:
Эту смесь охлаждают в холодильнике до 50°С и направляют на гидролиз, при котором протекают такие реакции:
Моноэтилсульфат, полученный в результате второй реакции, подвергают дальнейшему разложению с образованием еще одной молекулы спирта.
Прямая гидратация этилена
Технологическая схема производства этилового спирта способом прямой гидратации этилена представлена ниже.
Рисунок 4 – Технологическая схема прямой гидратации этилена при производстве этилового спиртаСырьем для способа прямой гидратации служит газ с высоким содержанием этилена (94-96%). Этилен сжимают компрессором до 8-9 КПа. Сжатый этилен смешивают с водяным паром в определённых соотношениях. Взаимодействие этилена с водяным паром производят в контактном аппарате – гидрататоре, представляющим собой вертикальную стальную полую цилиндрическую колонну, в которой находится катализатор (фосфорная кислота, нанесенная на алюмосиликат).
Смесь этилена и водяного пара при 280-300°С под давлением около 8,0 КПа подают в гидрататор, в котором поддерживают такие же параметры.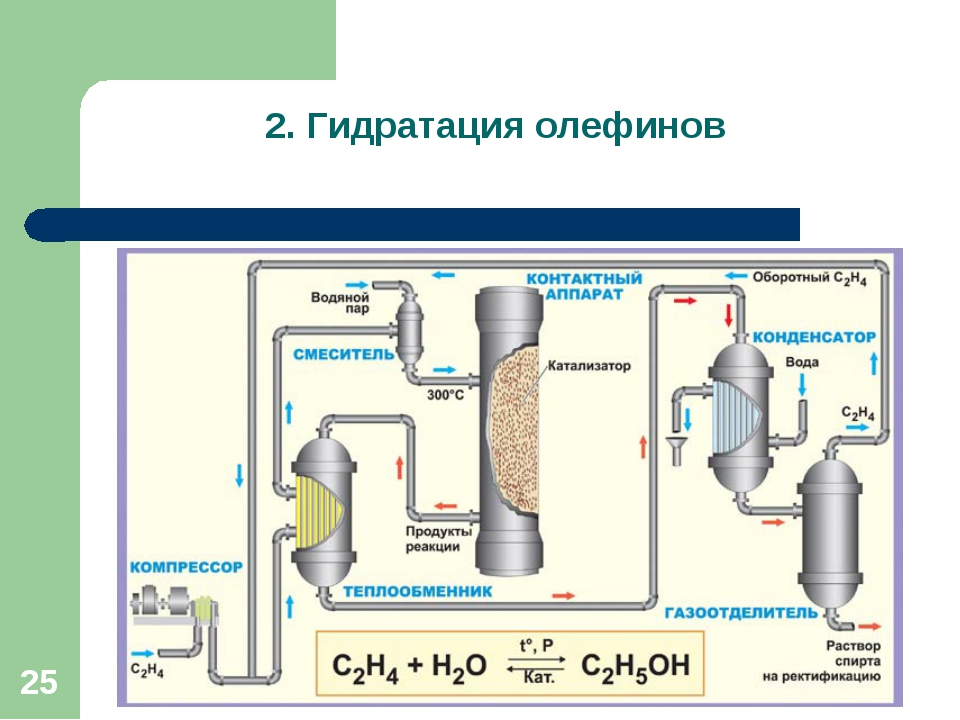 При взаимодействии этилена с водяным паром, кроме основной реакции образования этилового спирта, протекают побочные реакции, в результате которых получаются диэтиловый эфир, уксусный альдегид и продукты полимеризации этилена. Продукты синтеза уносят из гидрататора небольшое количество фосфорной кислоты, которая может в дальнейшем оказывать коррозийное действие на аппаратуру и трубопроводы. Чтобы избежать этого, кислоту, содержащуюся в продуктах синтеза, нейтрализуют щелочью. Продукты синтеза после нейтрализации пропускают через солеотделитель, а затем охлаждают в теплообменнике и производят конденсацию водно-спиртовых паров. Получают смесь водно-спиртовой жидкости и непрореагировавшего этилена. Непрореагировавший этилен отделяют от жидкости в сепараторе. Он представляет собой вертикальный цилиндр, в котором установлены перегородки, резко изменяющие скорость и направление газового потока. Этилен из сепаратора отводят во всасывающую линию циркуляционного компрессора и направляют на смешение со свежим этиленом.
При взаимодействии этилена с водяным паром, кроме основной реакции образования этилового спирта, протекают побочные реакции, в результате которых получаются диэтиловый эфир, уксусный альдегид и продукты полимеризации этилена. Продукты синтеза уносят из гидрататора небольшое количество фосфорной кислоты, которая может в дальнейшем оказывать коррозийное действие на аппаратуру и трубопроводы. Чтобы избежать этого, кислоту, содержащуюся в продуктах синтеза, нейтрализуют щелочью. Продукты синтеза после нейтрализации пропускают через солеотделитель, а затем охлаждают в теплообменнике и производят конденсацию водно-спиртовых паров. Получают смесь водно-спиртовой жидкости и непрореагировавшего этилена. Непрореагировавший этилен отделяют от жидкости в сепараторе. Он представляет собой вертикальный цилиндр, в котором установлены перегородки, резко изменяющие скорость и направление газового потока. Этилен из сепаратора отводят во всасывающую линию циркуляционного компрессора и направляют на смешение со свежим этиленом. Водно-спиртовой раствор, вытекающий из сепаратора, содержит 18,5-19% об. спирта. Его концентрируют в отпарной колонне и в виде паров направляют для очистки в ректификационную колонну. Спирт получают крепостью 90,5% об. На заводах синтетического спирта применяется способ прямой гидратации этилена.
Водно-спиртовой раствор, вытекающий из сепаратора, содержит 18,5-19% об. спирта. Его концентрируют в отпарной колонне и в виде паров направляют для очистки в ректификационную колонну. Спирт получают крепостью 90,5% об. На заводах синтетического спирта применяется способ прямой гидратации этилена.
Производство синтетического спирта, независимо от способа его получения, значительно более эффективно, чем производство спирта из пищевого сырья. Для получения 1 т этилового спирта из картофеля или зерна необходимо затратить 160-200 чел -дней , из газов нефтепереработки только 10 чел -дней . Себестоимость синтетического спирта примерно в четыре раза меньше себестоимости спирта из пищевого сырья.
Спирт из опилок в домашних условиях: технология производста биотоплива
Ограниченность запасов добываемых топливных ресурсов подталкивает человечество к поиску, выявлению и освоению альтернативных способов получения энергии. Наиболее перспективным направлением подобных изысканий становится получение замены газу и нефти из возобновляемых источников растительного происхождения. После биологической или термохимической обработки растительной массы получают биотопливо.
После биологической или термохимической обработки растительной массы получают биотопливо.
Одним из вариантов жидкого биотоплива является спирт из опилок. Такой способ применения отходов деревообработки – опилки – заменяет картофель и зерно при производстве спиртов. Для использования в получении спирта, 1 кг опилок равносилен 1 кг картофеля или 0,3 кг зерна. Очевидно, что такая замена сырья помогает сберечь в государственных масштабах огромные объемы пищевого сырья и освободить дополнительные площади для сельскохозяйственного производства.
Как сделать спирт из опилок
«Строительным» материалом растительных клеток дерева является твердая волокнистая клетчатка (целлюлоза). Ее содержание в растениях доходит до 70%. В клетчатке много полисахаридов, которые при определенном химическом воздействии переходят в глюкозу. Из последней после сбраживания и перегонки получают спирт. Значит, биомасса мелких опилок и древесной стружки из отходов превращается в ценное сырье для производства спирта – этанола или бутанола.
- Гидролиз измельченной древесной биомассы и последующее сбраживание полученного продукта с дрожжами. Это наиболее распространенный метод переработки опилок.
- Газификация древесины методом пиролиза и дальнейшее сбраживание образованного газа в спирт.
- Синтез газа при пиролизном разложении клетчатки опилок. Затем из газа получают метанол, который в результате реакции гомологенизации преобразуется в этанол.
Промышленное получение спирта
Производство спирта из древесных опилок на промышленной основе осуществляется двумя способами гидролиза. При любом из них опилки не требуют дополнительной подготовки, кроме максимально возможного измельчения.В первом случае опилки в вертикальном бункере заливаются концентрированным (40%) раствором серной кислоты. Массовые объемы опилок и раствора равны. Смесь герметично закрытого бункера доводят до температуры 220-250°С и регулярно перемешивая выдерживают от 1 ч до 1 ч 20 мин. Затем полученные при гидролизе растворы глюкозы и серной кислоты сцеживают. К слитой в другую емкость жидкости добавляют раствор мела для нейтрализации кислоты. Последующей фильтрацией отделяют раствор глюкозы от отходов.
Массовые объемы опилок и раствора равны. Смесь герметично закрытого бункера доводят до температуры 220-250°С и регулярно перемешивая выдерживают от 1 ч до 1 ч 20 мин. Затем полученные при гидролизе растворы глюкозы и серной кислоты сцеживают. К слитой в другую емкость жидкости добавляют раствор мела для нейтрализации кислоты. Последующей фильтрацией отделяют раствор глюкозы от отходов.
Метод не стал распространенным из-за высокой стоимости и повышенных требований к материалу применяемого оборудования, который должен выдерживать агрессивное воздействие концентрированной серной кислоты.
При втором, менее затратном, способе применяется слабый (0,5-1,0%) раствор кислоты. Процесс протекает 1,2-1,5 ч при температурах 160-190°С и давлении в бункере до 15 атм.
Общие моменты для рассмотренных методов гидролиза:
- Рабочие емкости изнутри выкладывают кислотоупорным материалом для исключения контакта кислоты с металлом.
- Загруженные в бункеры вещества нагревают с помощью подачи внутрь горячего пара.
 Выпускным клапаном для пара регулируют внутреннее давление рабочей емкости.
Выпускным клапаном для пара регулируют внутреннее давление рабочей емкости. - Гидролиз с успехом применяют для переработки опилок любых пород древесины.
- Полученная после химической реакции глюкоза используется для дальнейшего изготовления спирта, метанола, сахара.
- Рабочий процесс сопровождается выделением опасных газов. Для безопасности работников производственные помещения обязаны проветриваться, а сам персонал работает в специальных защитных респираторах и очках.
При производстве биотоплива из древесных опилок, получают побочные вещества, способные принести дополнительную прибыль:
- Лигнин – связующее вещество для получения пеллет и евродров из опилок и других измельченных материалов, активный наполнитель для пластмасс и синтетических каучуков.
- Фурфурол – жидкость, используемая как антисептик для древесины и компонент противогрибковых медикаментов. Также применяется для очистки масел и нефти, производства пластмасс, получения гербицидов, синтетических волокон и красителей.
 Другие свойства и перспективные варианты применения фурфурола изучаются.
Другие свойства и перспективные варианты применения фурфурола изучаются. - Скипидар.
- Алебастр.
Очистка раствора глюкозы
Процесс очистки проходит поэтапно:
- Сепараторная механическая очистка. На данном этапе из раствора выделяют лигнин.
- Нейтрализация серной кислоты при помощи обработки известковым молоком.
- Разделение раствора глюкозы и карбонатов (нужны при получении алебастра) отстаиванием.
Домашний способ получения спирта
Получить спирт из опилок можно в домашних условиях, и также двумя способами.Первый способ не требует наличия сложного оборудования, но является очень долгим (иногда неудачным) процессом, занимающим до 2-х лет. Собранные в кучу опилки поливают водой и оставляют преть под пленкой. Внутри кучи начинает повышаться температура и создаются условия для активизации процесса гидролиза с последующим получением глюкозы из клетчатки опилок. Технология дальнейшего использования глюкозы для брожения и получения спирта общеизвестна.
Главный недостаток естественного гидролиза – зависимость скорости протекания химических реакций от окружающей температуры, при значительном понижении которой процесс полностью прекращается. Также не исключается вероятность перерождения ожидаемого процесса в нежелательное гниение.
Второй способ копирует заводскую технологию. Сначала самостоятельно или на заказ делают уменьшенные аналоги промышленных установок. В качестве материала для них используют только нержавеющую сталь. Она стойкая к агрессивному воздействию слабого кислотного раствора.
Содержимое установки нагревают с помощью змеевика с циркулирующим разогретым паром (может применяться воздух) или на открытом огне. Регулируя внутреннее давление рабочей камеры, запускают процесс гидролиза. Рекомендуемые значения для эффективности протекания химической реакции – 7-10 атм.
Далее, как и при заводском производстве, из раствора механическим методом удаляется лигнин, затем нейтрализуется кислота и раствор отстаивается для оседания карбонатов. После очистки раствор глюкозы готов для последующего сбраживания.
После очистки раствор глюкозы готов для последующего сбраживания.
Брожение и перегонка
При последующем брожении глюкоза разлагается на спирт и углекислый газ. Для активизации процесса в домашних и промышленных условиях к раствору глюкозы добавляются дрожжи. Температура окружающей среды и порода древесины определяет время брожения – 5-14 суток. Окончание брожения определяется по значительному снижению интенсивности выделения газа. Визуально это характеризуется малым количеством поверхностной пены из пузырьков с углекислотой.
Последний технологический процесс – перегонка. Полученный жидкий материал нагревают до 70-80°С и выходящий пар охлаждают до выпадения спиртового конденсата. Для охлаждения пара, его пропускают через змеевик, который имеет водяное или воздушное наружное охлаждение.
После дальнейших 2-х-4-х перегонок готового продукта его крепость доводится до 80% и более. Теперь полученный спирт может называться биотопливом, на котором смогут эффективно работать двигатели внутреннего сгорания.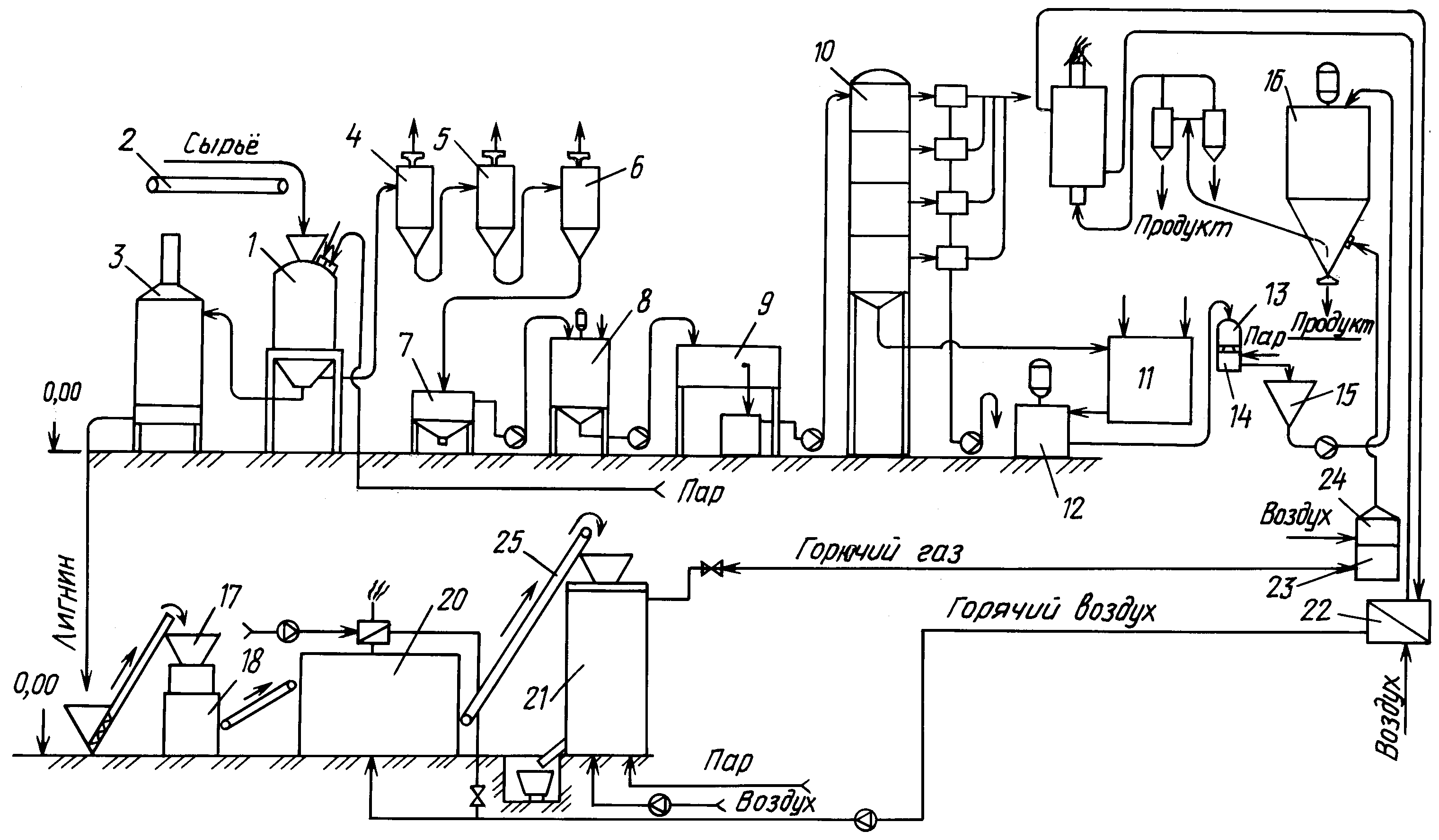
Остатки перегонки – скипидар и дрожжевая масса, пригодная для повторного сбраживания следующей партии рабочего раствора и приготовления кормовых дрожжей для животноводческих фермерских хозяйств.
Применение биотоплива
Биотопливо из опилок – биоэтанол – применяют для двигателей разнообразной техники. При такой замене бензину работа агрегата улучшается, увеличивается его мощность, снижается температура нагрева при нагрузках, увеличивается эксплуатационный ресурс поршней и клапанов, так как при сгорании спирта нет сажи и дыма.
При такой замене бензину работа агрегата улучшается, увеличивается его мощность, снижается температура нагрева при нагрузках, увеличивается эксплуатационный ресурс поршней и клапанов, так как при сгорании спирта нет сажи и дыма.
Этанол сделал возможным использование каминов в квартирах многоэтажек без привычного дымохода. Теплопотери при обогреве отсутствуют, дым и сажа не образуются, количество выделяемого углекислого газа незначительное.
ПРОИЗВОДСТВО ЭТИЛОВОГО СПИРТА ИЗ ДРЕВЕСИНЫ
Гидролиз полисахаридов растительной ткани в холодной воде практически не наблюдается. При повышении температуры воды выше 100° гидролиз полисахаридов протекает, но настолько медленно, что практического значения такой процесс не имеет. Удовлетворительные результаты получаются только при применении катализаторов, из которых производственное значение имеют лишь сильные минеральные кислоты: серная и реже соляная. Чем выше концентрация сильной кислоты в растворе и температура реакции, тем быстрее протекает гидролиз полисахаридов до моносахаридов. Однако присутствие таких катализаторов имеет и отрицательную сторону, так как они одновременно с реакцией гидролиза полисахаридов ускоряют и реакции распада моносахаридов, соответственно снижая этим их выход.
Однако присутствие таких катализаторов имеет и отрицательную сторону, так как они одновременно с реакцией гидролиза полисахаридов ускоряют и реакции распада моносахаридов, соответственно снижая этим их выход.
При распаде гексоз в этих условиях вначале образуется окси — метилфурфурол, который быстро разлагается далее с образованием конечных продуктов: левулиновой и муравьиной кислот. Пентозы в этих условиях превращаются в фурфурол.
В связи с этим, чтобы получить из полисахаридов растительной ткани моносахариды, необходимо обеспечить наиболее благоприятные условия для реакции гидролиза и максимально сократить возможности дальнейшего распада образующихся моносахаридов.
В этом заключается задача, которую решают исследователи и производственники при выборе оптимальных режимов гидролиза.
Из большого числа возможных вариантов концентрации кислоты и температуры реакции в настоящее время практически применяются только два: гидролиз разбавленными кислотами и гидролиз концентрированными кислотами. При гидролизе разбавленными кислотами температура реакции обычно составляет 160—190° и концентрация катализатора в водном растворе колеблется от 0,3 до 0,7% (h3S04, НС1).
При гидролизе разбавленными кислотами температура реакции обычно составляет 160—190° и концентрация катализатора в водном растворе колеблется от 0,3 до 0,7% (h3S04, НС1).
Реакцию проводят в автоклавах под давлением 10—15 атм. При гидролизе концентрированными кислотами концентрация серной кислоты обычно составляет 70—80%, а соляной 37—42%. Температура реакции в этих условиях 15—40°.
Снизить потери моносахаридов легче при гидролизе концентрированными кислотами, вследствие чего выход сахара при этом методе может достигать почти теоретически возможного, т, е. 650—750 кг из 1 т абсолютно сухого растительного сырья.
При гидролизе разбавленными кислотами снизить потери моносахаридов вследствие их разложения значительно труднее и поэтому практически выход моносахаридов в этом случае обычно не превышает 450—500 кг из 1 г сухого сырья.
Ввиду малых потерь сахара при гидролизе концентрированными кислотами получающиеся водные растворы моносахаридов — гидролизаты отличаются повышенной чистотой, что имеет большое значение при их последующей переработке.

Серьезным недостатком методов гидролиза концентрированными кислотами до последнего времени был большой расход минеральной кислоты на тонну получаемого сахара, что приво дило к необходимости регенерации части кислоты или использования ее в других производствах; это осложняло и удорожало строительство и эксплуатацию таких заводов.
Большие трудности возникали также при подборе для аппаратуры материалов, стойких в агрессивных средах. По этой причине основная масса действующих в настоящее время гидролизных заводов была построена по методу гидролиза разбавленной серной кислотой.
Первый опытный гидролизно-спиртовый завод в СССР был пущен в январе 1934 г. в г. Череповце. Исходные показатели и технический проект этого завода были разработаны кафедрой гидролизных производств Ленинградской лесотехнической академии в 1931 —1933 гг. На основе данных эксплуатации опытного завода было начато строительство в СССР промышленных гид — ролизно-спиртовых заводов. Первый промышленный гидролизно — спиртовый завод был пущен в Ленинграде в декабре 1935 г.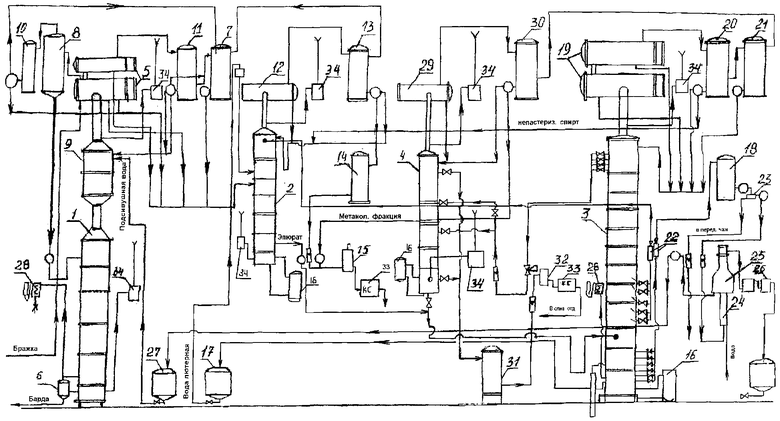 Вслед за этим заводом в период 1936—1938 гг. вошли в строй Бобруйский, Хорский и Архангельский гидролизно-спиртовые заводы. Во время второй мировой войны и после нее было построено много больших заводов в Сибири и на Урале. В настоящее время проектная мощность этих заводов в результате совершенствования технологии перекрыта в 1,5—2 раза.
Вслед за этим заводом в период 1936—1938 гг. вошли в строй Бобруйский, Хорский и Архангельский гидролизно-спиртовые заводы. Во время второй мировой войны и после нее было построено много больших заводов в Сибири и на Урале. В настоящее время проектная мощность этих заводов в результате совершенствования технологии перекрыта в 1,5—2 раза.
Основным сырьем для этих заводов является хвойная древесина в виде опилок и щепы, поступающая с соседних лесопильных заводов, где ее получают путем измельчения в рубительных машинах отходов лесопиления — горбыля и рейки. В отдельных случаях измельчают и хвойные дрова.
Схема получения моносахаридов на таких заводах представлена на рис. 76.
Измельченная хвойная древесина со склада сырья по транспортеру 1 поступает в направляющую воронку 2 и далее в горло-
Вину гидролизаппарата 3. Это вертикальный стальной цилиндр с верхним и нижним конусами и горловинами. Внутреннюю поверхность такого гидролизаппарата покрывают кислотоупорными керамическими или графитовыми плитками или кирпичом, укрепленным на слое бетона толщиной 80—100 мм. Швы между плитками заполняются кислотоупорной замазкой. Верхняя и нижняя горловины гидролизаппарата с внутренней стороны защищены от действия горячей разбавленной серной кислоты слоем кислотоупорной бронзы. Полезный объем таких гидролизаппа — ратов обычно составляет 30—37 At3, но иногда применяются также гидролизаппараты объемом 18, 50 и 70 м3. Внутренний диаметр таких гидролизаппаратов составляет около 1,5, а высота 7—13 м. В верхний конус гидролизаппарата во время гидролиза по трубе 5 подается нагретая до 160—200° разбавленная серная кислота.
Внутреннюю поверхность такого гидролизаппарата покрывают кислотоупорными керамическими или графитовыми плитками или кирпичом, укрепленным на слое бетона толщиной 80—100 мм. Швы между плитками заполняются кислотоупорной замазкой. Верхняя и нижняя горловины гидролизаппарата с внутренней стороны защищены от действия горячей разбавленной серной кислоты слоем кислотоупорной бронзы. Полезный объем таких гидролизаппа — ратов обычно составляет 30—37 At3, но иногда применяются также гидролизаппараты объемом 18, 50 и 70 м3. Внутренний диаметр таких гидролизаппаратов составляет около 1,5, а высота 7—13 м. В верхний конус гидролизаппарата во время гидролиза по трубе 5 подается нагретая до 160—200° разбавленная серная кислота.
В нижнем конусе установлен фильтр 4 для отбора полученного гидролизата. Гидролиз в таких аппаратах производится периодически.
Как уже указывалось выше, гидролизаппарат загружают измельченным сырьем через направляющую воронку. При загрузке сырья через трубу 5 поступает нагретая до 70—90° разбавленная серная кислота, которая смачивает сырье, способствуя его уплотнению. При таком методе загрузки в 1 м3 гидролизаппарата помещается около 135 кг опилок или 145—155 кг Щепы, в пересчете на абсолютно сухую древесину. По окончании загрузки содержимое гидролизаппарата подогревается острым паром, поступающим в нижний конус его. Как только будет достигнута температура 150—170°, в гидролизаппарат по трубе 5 начинает поступать 0,5—0,7’%-пая серная кислота, нагретая до 170—200°. Одновременно образующийся гидролизат через фильтр 4 начинает выводиться в испаритель б. Реакция гидролиза в гидролизаппарате продолжается от 1 до 3 часов. Чем короче время гидролиза, тем выше температура и давление в гидролизаппарате.
При загрузке сырья через трубу 5 поступает нагретая до 70—90° разбавленная серная кислота, которая смачивает сырье, способствуя его уплотнению. При таком методе загрузки в 1 м3 гидролизаппарата помещается около 135 кг опилок или 145—155 кг Щепы, в пересчете на абсолютно сухую древесину. По окончании загрузки содержимое гидролизаппарата подогревается острым паром, поступающим в нижний конус его. Как только будет достигнута температура 150—170°, в гидролизаппарат по трубе 5 начинает поступать 0,5—0,7’%-пая серная кислота, нагретая до 170—200°. Одновременно образующийся гидролизат через фильтр 4 начинает выводиться в испаритель б. Реакция гидролиза в гидролизаппарате продолжается от 1 до 3 часов. Чем короче время гидролиза, тем выше температура и давление в гидролизаппарате.
В процессе гидролиза полисахариды древесины переходят в соответствующие моносахариды, растворяющиеся в горячей разбавленной кислоте. Для предохранения этих моносахаридов от разложения при высокой температуре содержащий их гидролизат непрерывно в течение всей варки выводят через фильтр 4 И быстро охлаждают в испарителе 6. Так как по условиям процесса гидролизуемое растительное сырье. в гидролизаппарат» все время должно быть залито жидкостью, заданный уровень е поддерживается горячей кислотой, поступающей по трубе 5,
Так как по условиям процесса гидролизуемое растительное сырье. в гидролизаппарат» все время должно быть залито жидкостью, заданный уровень е поддерживается горячей кислотой, поступающей по трубе 5,
Такой метод работы носит название перколяция. Чем быст рее идет перколяция, т. е. чем быстрее через гидролизаппарат протекает горячая кислота, тем быстрее образующийся сахар выводится из реакционного пространства и тем меньше он разлагается. С другой стороны, чем быстрее идет перколяция, тем больше расходуется на варку горячей кислоты и тем меньше получается концентрация сахара в гидролизате и соответственно больше расход пара и кислоты на варку.
Практически для получения достаточно высоких выходов сахара (при экономически приемлемой концентрации его в гидролизате) приходится выбирать некоторые средние условия пер — коляции. Обычно останавливаются на выходе сахара в 45—50% от веса абсолютно сухой древесины при концентрации сахара в гидролизате 3,5—3,7 % — Эти оптимальные условия реакции соответствуют отбору через нижний фильтр из гидролизаппара — та 12—15 м3 гидролизата на 1 т абсолютно сухой древесины, загруженной в гидролизаппарат. бе 21. Для этой цели быстро открывают клапан 20, соединяющий внутреннее пространство гидролизаппарата с циклоном 22. Благодаря быстрому снижению давления между кусочками лигнина содержащаяся в нем перегретая вода мгновенно вскипает, образуя большие объемы пара. Последний рвет лигнин и увлекает его в виде взвеси по трубе 21 в циклон 22. Труба 21 подходит к циклону по касательной, благодаря чему струя пара с лигнином, врываясь в циклон, движется вдоль стенок, совер — шая вращательное движение. Лигнин центробежной силой отбрасывается к боковым стенкам и, теряя скорость, падает на дно циклона. Освобожденный от лигнина пар через центральную трубу 23 выбрасывается в атмосферу.
бе 21. Для этой цели быстро открывают клапан 20, соединяющий внутреннее пространство гидролизаппарата с циклоном 22. Благодаря быстрому снижению давления между кусочками лигнина содержащаяся в нем перегретая вода мгновенно вскипает, образуя большие объемы пара. Последний рвет лигнин и увлекает его в виде взвеси по трубе 21 в циклон 22. Труба 21 подходит к циклону по касательной, благодаря чему струя пара с лигнином, врываясь в циклон, движется вдоль стенок, совер — шая вращательное движение. Лигнин центробежной силой отбрасывается к боковым стенкам и, теряя скорость, падает на дно циклона. Освобожденный от лигнина пар через центральную трубу 23 выбрасывается в атмосферу.
Циклон 22 обычно представляет собой вертикальный сталь-‘ ной цилиндр объемом около 100 м3, снабженный боковой дверцей 31 и вращающейся мешалкой 25, которая помогает при выгрузке лигнина со дна циклона на ленточный или скребковый транспортер 24.
Для предохранения от коррозии внутренняя поверхность циклонов иногда защищается слоем кислотоупорного бетона Как уже указывалось выше, в процессе перколяции в верхний конус гидролизаппарата подается нагретая разбавленная серная кислота. Ее приготовляют путем смешивания в кислотоупорном смесителе 17 перегретой воды, подаваемой по трубе 28, с холодной концентрированной серной кислотой, поступающей из мерного бачка 19 через поршневой кислотный насос 18.
Поскольку холодная концентрированная серная кислота слабо корродирует железо и чугун, эти металлы широко используют для изготовления баков, насосов и трубопроводов, предназначенных для ее хранения и транспортировки к смесителю. Аналогичные материалы применяются и для подвода перегретой йоды к смесителю. Для защиты стенок смесителя от коррозии Применяют фосфористую бронзу, графит или пластическую массу — фторопласт 4. Последние два используются для внутренней футеровки смесителей и дают наилучшие результаты.
Готовый гндролизат из гидролизаппарата поступает в испаритель 6 высокого давления. Это — стальной сосуд, работающий под давлением и футерованный внутри керамическими плитками, как и гидролизаппарат. В верхней части испарителя емкостью 6—8 ж3 имеется крышка. В испарителе поддерживается давление на 4—5 атм ниже, чем в гидролизаппарате. Благодаря этому попадающий в него гидролизат мгновенно вскипает, частично испаряясь, и охлаждается до 130—140°. Образующийся пар отделяется от капель гидролизата и по трубе 10 поступает в решофер (теплообменник) 11, где конденсируется. Частично охлажденный гидролизат из испарителя 6 по трубе 7 поступает в испаритель 8 низкого давления, где охлаждается до 105—110° в результате вскипания при более низком давлении, обычно не превышающем одной атмосферы. Образующийся в этом испарителе пар по трубе 14 подается во второй решофер 13, где также конденсируется. Конденсаты из решоферов 11 и 13 содержат 0,2—0,3% фурфурола и используются для его выделения на специальных установках, которые будут рассмотрены ниже.
Тепло, содержащееся в паре, который выходит из испарителей 6 и 8, используется для нагрева воды, поступающей в смеситель 17. Для этой цели из бака 16 оборотной воды насосом 1Ь Теплую воду, полученную из ректификационного отделения гидролизного завода, подают в решофер низкого давления 13, где она нагревается с 60—80° до 100—110°. Затем по трубе 12 подогретая вода проходит решофер высокого давления 11, где паром при температуре 130—140° подогревается до 120—130°. Дальше температуру воды повышают до 180—200° в водогрейной колонне 27. Последняя представляет собой вертикальный стальной цилиндр с дном и верхней крышкой, рассчитанными на рабочее давление 13—15 атм.
Пар в водогрейную колонку подают по вертикальной трубе 26, на конце которой укреплены 30 горизонтальных дисков 2Ь. Пар из трубы 26 проходит через щели между отдельными дисками в колонну, заполненную водой. Последняя непрерывно подается в колонну через нижний штуцер, смешивается с паром, нагревается до заданной температуры и по трубе 28 поступает в смеситель 17.
Гидролизаппараты устанавливают на специальном фундаменте в ряд по 5—8 шт. На больших заводах число их удваивают и устанавливают их в два ряда. Трубопроводы для гидролизата изготовляют из красной меди или латуни. Арматура, состоящая из вентилей и клапанов, изготовляется из фосфористой или паспортной бронзы.
Описанный выше способ гидролиза является периодическим. В настоящее время испытываются новые конструкции гидролпз — аппаратов непрерывного действия, в которые при помощи специальных питателей непрерывно подается измельченная древесина, непрерывно удаляется лигнин и гидролизат.
Ведутся также работы по автоматизации гидролизаппаратов периодического действия. Это мероприятие позволяет более точно соблюдать заданный режим варки и одновременно облегчает труд варщиков.
Кислый гидролизат из испарителя низкого давления 8 (рис. 76) по трубе 9 подают в аппаратуру для его последующей переработки. Температура такого гидролизата 95—98°. В нем содержится (в %):
Серной кислоты. . . ……………………………………………………………………………………………….. 0,5 —0,7:
. . ……………………………………………………………………………………………….. 0,5 —0,7:
Гексоз (глюкоза, манноза, галактоза)………………………………………………………….. 2,5 —2,8;
Пентоз (ксилоза, арабиноза)…………………………………………………………………………. 0,8 —1,0;
Летучих органических кислот (муравьиная, уксусная) …………………………….. 0,24-0,30;
Нелетучих органических кислот (левулиновая) . . 0,2 —0,3;
Фурфурола………………………………………………………………………………………………………. 0,03—0,05;
Оксиметилфурфурола……………………………………………………………………………………. 0,13—0,16;
Метанола. ……………………………………………………………………………………………………….. 0,02-0,03
В гидролизатах присутствуют также коллоидные вещества (лигнин, декстрины), зольные вещества, терпены, смолы и т. д. Содержание моносахаридов в растительных гидролизатах при точных химических исследованиях устанавливают путем количественной бумажной хроматографии.
В заводских лабораториях при массовых экспрессных определениях Сахаров используется способность их в щелочной среде восстанавливать комплексные соединения окиси меди с образованием закиси меди:
2 Си (ОН)2 Си5 О + 2 Н2 О + 02.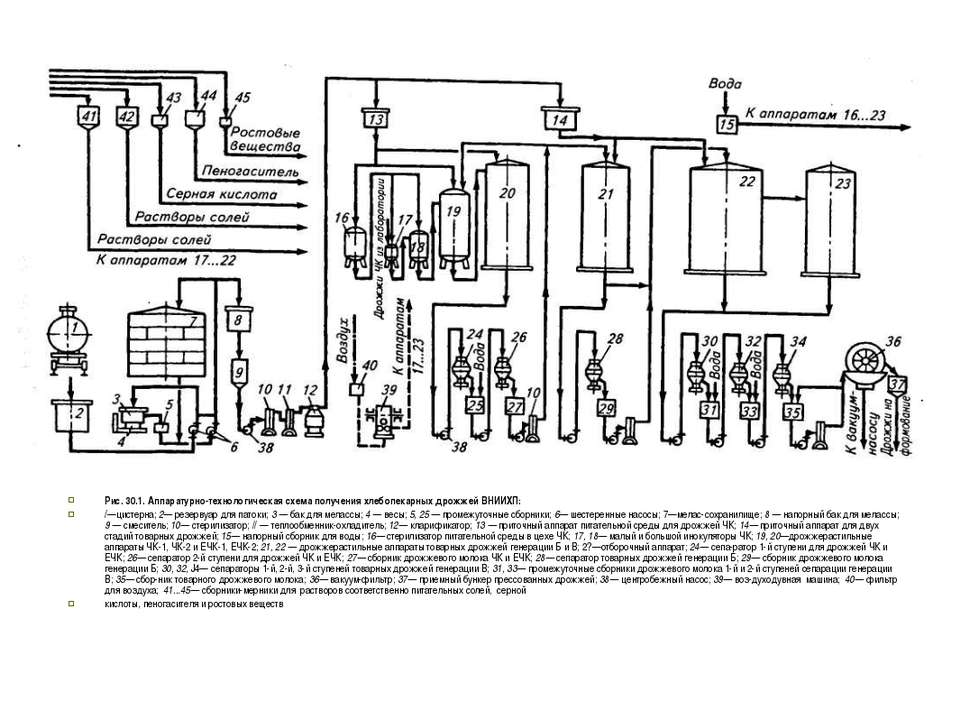
По количеству образующейся закиси меди вычисляется со — i-фжание моносахаридов в растворе.
Такой метод определения Сахаров является условным, так Как одновременно с моносахаридами окись меди восстанавливают в закись также фурфурол, оксиметилфурфурол, декстрины, коллоидный лигнин. Эти примеси мешают определению истинного содержания сахара в гидролизатах. Общая ошибка здесь достигает 5—8%. Поскольку поправка на эти примеси требует большой затраты труда, ее обычно не делают, а полученные сахара в отличие от моносахаридов называют редуцирующими веществами или сокращено РВ. В заводских условиях учет количества вырабатываемого сахара в гидролизате учитывают в тоннах РВ.
Для получения этилового спирта гексозы (глюкоза, манноза и галактоза) сбраживают спиртообразующими дрожжами — сахаромицетами или шизосахаромицетами.
Суммарное уравнение спиртового брожения гексоз
C(i Hf, 06 — 2 С2 НГ) ОН + 2 С02 Гексоза этиловый спирт
Показывает, что при этом процессе теоретически на каждые 100 кг сахара должно получаться 51,14 кг, или около 64 л 100%-ного этилового спирта и около 49 кг углекислоты.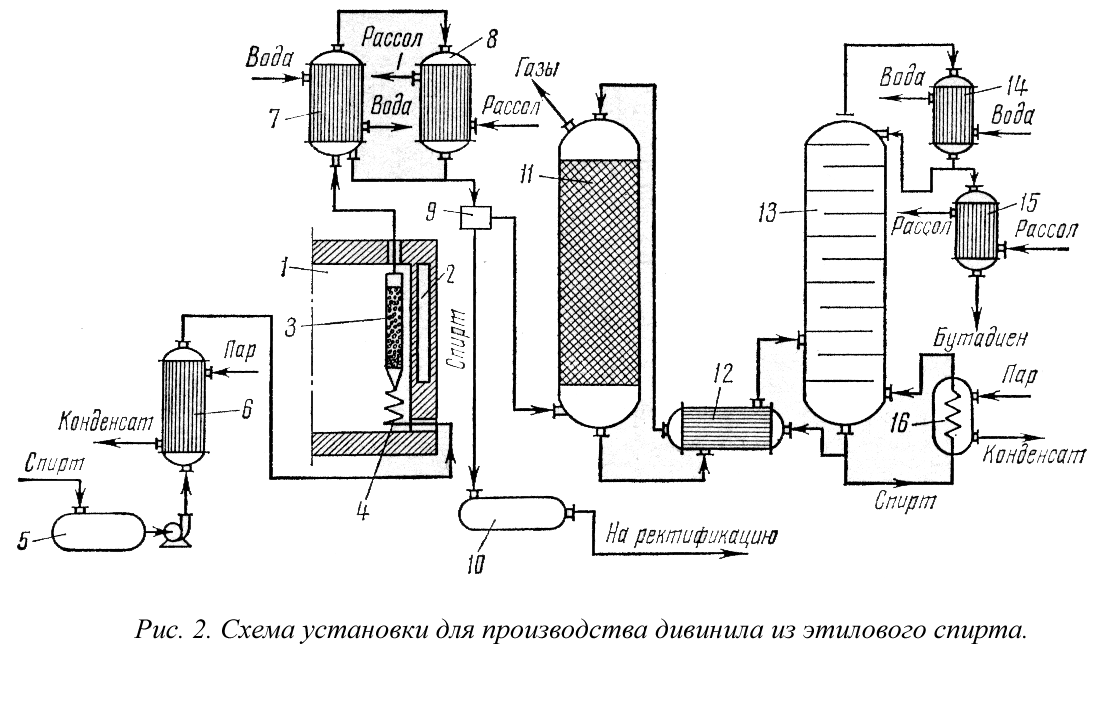
Таким образом, при спиртовом брожении гексоз получается почти в равных количествах два основных продукта: этанол и углекислота. Для осуществления этого процесса горячий кислый гидролизат должен быть подвергнут следующей обработке.:
1) нейтрализации; 2) освобождению от взвешенных твердых частиц; 3) охлаждению до 30°; 4) обогащению гидролизата необходимыми для жизнедеятельности дрожжей питательными веществами.
Кислый гидролизат имеет рН=1 —1,2. Среда, пригодная для брожения, должна иметь рН = 4,6—5,2. Чтобы придать гидро — лизату необходимую кислотность, содержащиеся в нем свободную серную и значительную часть органических кислот необходимо нейтрализовать. Если все кислоты, содержащиеся в гидролизате, условно выразить в серной кислоте, то ее концентрация составит около 1%. Остаточная кислотность гидролизата при рН = 4,6—5,2 составляет около 0,15%.
Поэтому для получения в гидролизате необходимой концентрации ионов водорода, в нем должно быть нейтрализовано 0,85% кислот. При этом полностью нейтрализуется свободная серная, муравьиная и часть уксусной. Остаются свободными левулиновая кислота и небольшая часть уксусной.
При этом полностью нейтрализуется свободная серная, муравьиная и часть уксусной. Остаются свободными левулиновая кислота и небольшая часть уксусной.
Нейтрализуют гидролизат известковым молоком, т. е. суспензией гидрата окиси кальция в воде с концентрацией 150—200 г СаО в литре.
Схема приготовления известкового молока представлена нм рис. 77.
Негашеную известь СаО непрерывно подают в загрузочную воронку вращающегося известегасительного барабана 34. Одновременно в барабан подают необходимое количество воды. При вращении барабана негашеная известь, связывая воду, переходит в гидрат окиси кальция. Последний диспергируется в воде, образуя суспензию. Не прореагировавшие куски извести отделяются в конце барабана от известкового молока и сбрасываются в вагонетку. Известковое молоко вместе с песком протекает по трубе в отделитель песка 35. Последний представляет собой горизонтально расположенное железное корыто с поперечными перегородками и продольным валом с лопастями.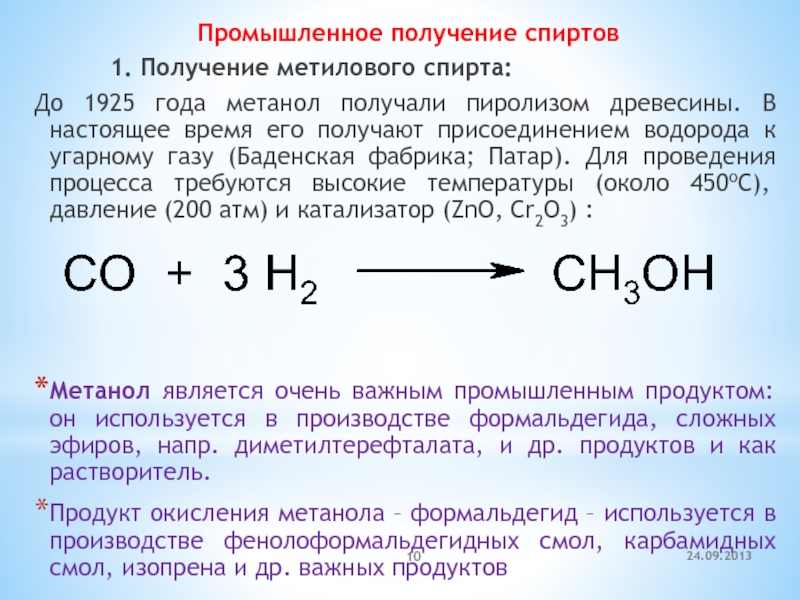
Известковое молоко в этом аппарате медленно течет справа налево и далее по трубе 36 сливается в сборник 2.
Песок медленно оседает между перегородками отделителя песка и при помощи медленно вращающихся лопаток удаляется из аппарата. Перед поступлением известкового молока в нейтрализатор его смешивают с заданным количеством сернокислого аммония, раствор которого поступает из бачка 37. При смешении известкового молока с сернокислым аммонием протекает реакция
Са (ОН)3 + (Nh5)2 S04-> CaS04 + 2 NH, ОН, в результате которой часть извести связывается серной кислотой сернокислого аммония и образуются кристаллы плохо растворимого двухводного сернокислого кальция CaS04-2h30. Одновременно образуется аммиак, остающийся в известковом молоке в растворенном состоянии.
Присутствующие в известковом молоке мелкие кристаллы гипса при последующей нейтрализации являются центрами кристаллизации образующегося гипса и предохраняют от образования пересыщенных растворов его в нейтрализованном гидролизате. Это мероприятие имеет важное значение при последующей отгонке спирта из бражки, так как пересыщенные растворы гипса в бражке вызывают гипсацию бражных колонн и быстро выводят их из строя. Такой метод работы получил название нейтрализации с направленной кристаллизацией гипса.
Это мероприятие имеет важное значение при последующей отгонке спирта из бражки, так как пересыщенные растворы гипса в бражке вызывают гипсацию бражных колонн и быстро выводят их из строя. Такой метод работы получил название нейтрализации с направленной кристаллизацией гипса.
Одновременно с известковым молоком в нейтрализатор 5 Подаются слабокислый водный экстракт суперфосфата из мерника-бачка 38.
Соли даются в нейтрализатор из расчета 0,3 кг сернокислого аммония и 0,3 кг суперфосфата на 1 м3 гидролизата.
Нейтрализатор 5 (емкостью 35—40 м3) представляет собой стальной бак, футерованный кислотоупорными керамическими плитками и снабженный вертикальными мешалками и тормозными лопатками, укрепленными неподвижно на стенках бака. Нейтрализация на гидролизных заводах ранее производилась периодически. В настоящее время она вытесняется более совершенной непрерывной нейтрализацией. На рис. 77 приведена последняя схема. Процесс осуществляется в двух последовательно соединенных нейтрализаторах 5 и 6, имеющих одинаковое устройство. Кислый гидролизат по трубе 1 непрерывно подается в первый нейтрализатор, куда одновременно поступают известковое молоко и питательные соли. Контроль за полнотой нейтрализации производят путем измерения концентрации ионов водорода при помощи потенциометра 3 с сурьмяным или стеклянным электродом 4. Потенциометр непрерывно записывает рН гидролизата и автоматически регулирует его в заданных пределах, посылая электрические импульсы реверсивному мотору, соединенному с запорной арматурой на трубе, подающей известковое молоко в первый нейтрализатор. В нейтрализаторах сравнительно быстро протекает реакция нейтрализации и относительно медленно — процесс кристаллизации гипса из пересыщенного раствора.
Процесс осуществляется в двух последовательно соединенных нейтрализаторах 5 и 6, имеющих одинаковое устройство. Кислый гидролизат по трубе 1 непрерывно подается в первый нейтрализатор, куда одновременно поступают известковое молоко и питательные соли. Контроль за полнотой нейтрализации производят путем измерения концентрации ионов водорода при помощи потенциометра 3 с сурьмяным или стеклянным электродом 4. Потенциометр непрерывно записывает рН гидролизата и автоматически регулирует его в заданных пределах, посылая электрические импульсы реверсивному мотору, соединенному с запорной арматурой на трубе, подающей известковое молоко в первый нейтрализатор. В нейтрализаторах сравнительно быстро протекает реакция нейтрализации и относительно медленно — процесс кристаллизации гипса из пересыщенного раствора.
Поэтому скорость протекания жидкости через нейтрализа — ционную установку обусловлена вторым процессом, требующим для своего окончания 30—40 мин.
По истечении этого времени нейтрализованный гидролизат, называемый «нейтрализатом», поступает в отстойник 7 полунепрерывного или непрерывного действия.
Полунепрерывный процесс состоит в том, что нейтрализат протекает через отстойник непрерывно, а оседающий на дно его гипс удаляется периодически, по мере накопления.
При непрерывной работе отстойника все операции производятся непрерывно. Перед спуском в канализацию шлам 8 в приемнике дополнительно промывается водой. Последний способ из-за некоторых производственных трудностей еще не получил широкого распространения.
Гипсовый шлам из отстойника обычно состоит наполовину из двухводного сернокислого кальция и наполовину из лигнина и гуминовых веществ, осевших из гидролизата. На некоторых гидролизных заводах гипсовый шлам обезвоживают, высушивают и обжигают, превращая в строительный алебастр. Обезвоживают на барабанных вакуум-фильтрах, а высушивают и обжигают во вращающихся барабанных печах, обогреваемых топочными газами.
Нейтрализат, освобожденный от взвешенных частиц, перед брожением охлаждается в холодильнике 10 (рис. 77) с 85 до 30°. Для этой цели обычно применяются спиральные или пластинчатые теплообменники, отличающиеся высоким коэффициентом теплопередачи и небольшими габаритами. При охлаждении из нейтрализата выделяются смолообразные вещества, которые оседают на стенках теплообменников и постепенно загрязняют их. Для чистки теплообменники периодически отключают и промывают 2—4%-ным горячим водным раствором едкого натра, который растворяет смолообразные и гуминовые вещества.
При охлаждении из нейтрализата выделяются смолообразные вещества, которые оседают на стенках теплообменников и постепенно загрязняют их. Для чистки теплообменники периодически отключают и промывают 2—4%-ным горячим водным раствором едкого натра, который растворяет смолообразные и гуминовые вещества.
Нейтрализованный, очищенный и охлажденный гидролизат.
Содержащий необходимые питательные соли, носит название древесного сусла.
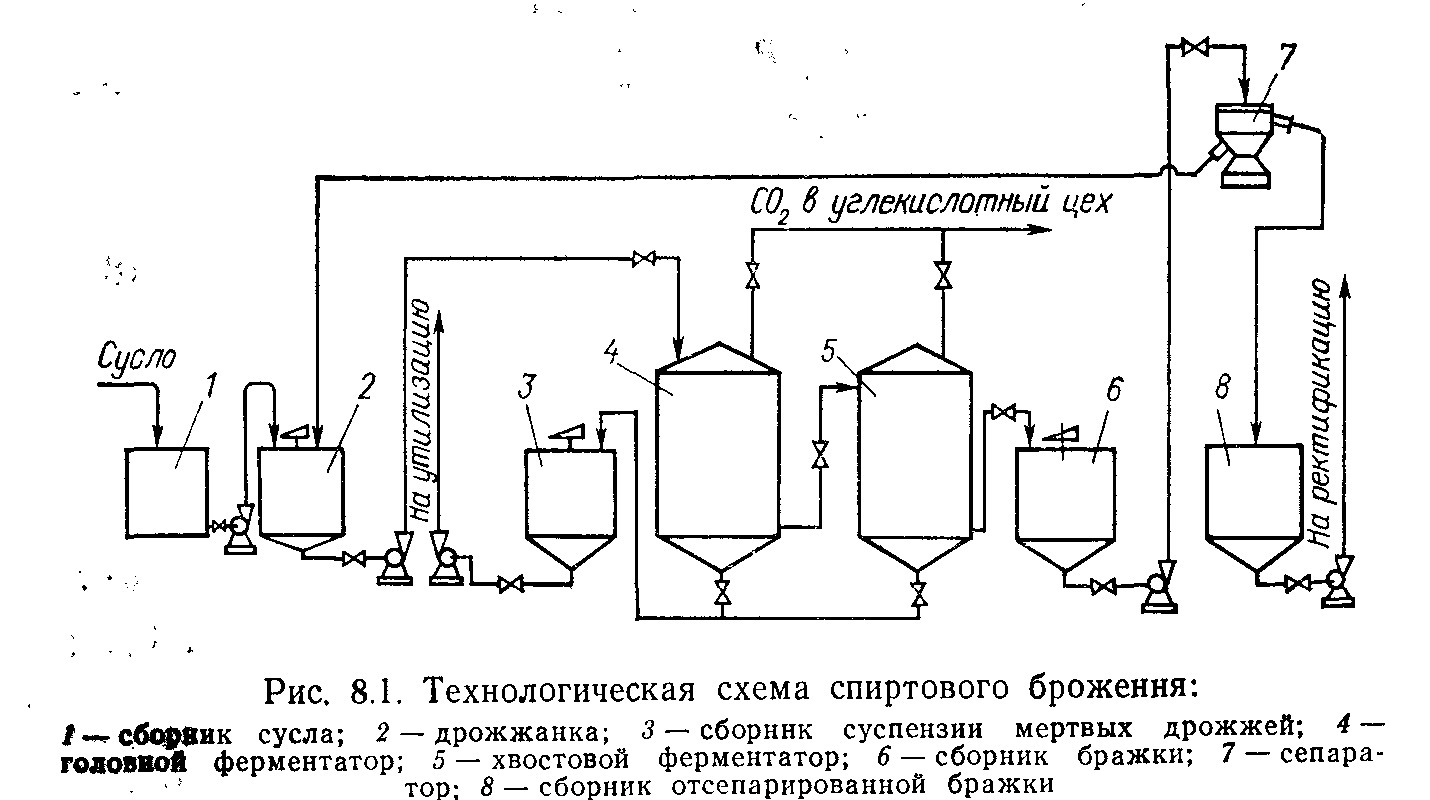
Сбраживают древесное сусло специальными акклиматизированными в этой среде спнртообразующими дрожжами. Брожение идет по непрерывному методу в батарее последовательно соединенных бродильных чанов 11 и 12.
Дрожжевая суспензия, содержащая около 80—100 г прессованных[1] дрожжей в литре, подается непрерывным потоком по трубе 15 в дрожжанку 44 и затем в верхнюю часть первого, или головного, бродильного чана 11. В дрожжанку одновременно’ с дрожжевой суспензией подается охлажденное древесное сусло. На каждый кубометр дрожжевой суспензии в бродильный чан поступает 8—10 м3 сусла.
Дрожжинки, содержащиеся в среде гексозных Сахаров, при помощи системы ферментов расщепляют сахара, образуя этиловый спирт и углекислоту. Этиловый спирт переходит в окружающую жидкость, а углекислый газ выделяется на поверхности дрожжинок в виде маленьких пузырьков, которые постепенно’ увеличиваются в объеме, затем постепенно всплывают на поверхность чана, увлекая приставшие к ним дрожжинки.
При соприкосновении с поверхностью пузырьки углекислоты лопаются, а дрожжинки, имеющие удельный вес 1,1, т. е. больший, чем у сусла (1,025), опускаются вниз, пока снова не будут подняты углекислотой на поверхность. Непрерывное движение дрожжинок вверх и вниз способствует перемещению потоков жидкости в бродильном чане, создавая перемешивание или «брожение» жидкости. Выделяющаяся на поверхности жидкости углекислота из бродильных чанов по трубе 13 поступает на установку для получения жидкой или твердой углекислоты, используется для получения химических продуктов (например, мочевины) или выпускается в атмосферу.
Частично сброженное древесное сусло вместе с дрожжами передается из головного бродильного чана в хвостовой чан 12, Где брожение и заканчивается. Поскольку концентрация сахарз в хвостовом чане небольшая, брожение в нем идет менее интенсивно, и часть дрожжей, не успевая образовать пузырьки углекислоты, оседает на дно чана. Чтобы не допустить этого, в хвостовом чане устраивают часто принудительное перемешивание жидкости мешалками или центробежными насосами.
Бродящая или сброженная жидкость называется бражкой. По окончании брожения бражка передается в сепаратор 14, работающий по принципу центрифуги. Попадающая в него бражка вместе со взвешенными в ней дрожжами начинает вращаться со скоростью 4500—6000 оборотов в минуту. Центробежная сила вследствие разности удельных весов бражки и дрожжей разделяет их. Сепаратор делит жидкость на два потока: больший, не содержащий дрожжей, поступает в воронку 16 и меньший, содержащий дрожжи, поступает через воронку в трубу 15. Обычно первый поток в 8—10 раз больше, чем второй. По трубе 15 дрожжевая суспензия возвращается в головной бродильный чан 11 Через дрожжанку 44. Сброшенное и освобожденное от дрожжей сусло собирается в промежуточном сборнике бражки 17.
Обычно первый поток в 8—10 раз больше, чем второй. По трубе 15 дрожжевая суспензия возвращается в головной бродильный чан 11 Через дрожжанку 44. Сброшенное и освобожденное от дрожжей сусло собирается в промежуточном сборнике бражки 17.
При помощи сепараторов дрожжи постоянно циркулируют в замкнутой системе бродильной установки. Производительность сепараторов 10—35 м3/час.
Во время брожения и особенно при сепарации часть содержащихся в древесном сусле гуминовых коллоидов коагулируется, образуя тяжелые хлопья, медленно оседающие на дно бродильных чанов. В днищах чанов устроены штуцеры, через которые осадок периодически спускается в канализацию.
Как уже указывалось выше, теоретический выход спирта из 100 кг сброженных гексоз составляет 64 л. Однако практически вследствие образования за счет Сахаров побочных продуктов (глицерин, уксусный альдегид, янтарная кислота и т. д.), а также из-за присутствия в сусле вредных для дрожжей примесей выход спирта составляет 54—56 л.
Для получения хороших выходов спирта необходимо все время дрожжи поддерживать в активном состоянии. Для этого следует тщательно выдерживать заданную температуру брожения, концентрацию водородных ионов, необходимую чистоту сусла и оставлять в бражке перед поступлением ее на сепаратор небольшое количество гексоз, так называемый «недоброд» (обычно не более 0,1 %’ сахара в растворе). Благодаря наличию недоброда дрожжи все время остаются в активной форме.
Периодически гидролизный завод останавливают на планово — предупредительный или капитальный ремонт. В это время дрожжи следует сохранять в живом виде. Для этого суспензию дрожжей при помощи сепараторов сгущают и заливают холодным древесным суслом. При низкой температуре брожение резко замедляется и дрожжи потребляют значительно меньше сахара.
Бродильные чаны емкостью 100—200 м3 обычно изготовляются из листовой стали или, реже, из железобетона. Продолжительность брожения зависит от концентрации дрожжей и составляет от 6 до 10 часов. Необходимо следить за чистотой производственной культуры дрожжей и предохранять ее от инфицирования посторонними вредными микроорганизмами. Для этой цели все оборудование необходимо содержать в чистоте и периодически подвергать стерилизации. Наиболее простым способом стерилизации является пропарка всего оборудования и особенно трубопроводов и насосов острым паром.
Необходимо следить за чистотой производственной культуры дрожжей и предохранять ее от инфицирования посторонними вредными микроорганизмами. Для этой цели все оборудование необходимо содержать в чистоте и периодически подвергать стерилизации. Наиболее простым способом стерилизации является пропарка всего оборудования и особенно трубопроводов и насосов острым паром.
По окончании брожения и отделения дрожжей спиртовая бражка содержит от 1,2 до 1,6% этилового спирта и около 1% пентозных Сахаров.
Выделяют спирт из бражки, очищают и укрепляют его в трехколонном брагоректификационном аппарате, состоящем из браж — ной 18, ректификационной 22 и метанольной 28 колонн (рис.77).
Бражка из сборника 17 насосом подается через теплообменник 41 на питающую тарелку бражной колонны 18. Стекая по тарелкам исчерпывающей части бражной колонны вниз, бражка встречает на своем пути поднимающийся вверх пар. Последний, постепенно обогащаясь спиртом, переходит в верхнюю, укрепляющую часть колонны.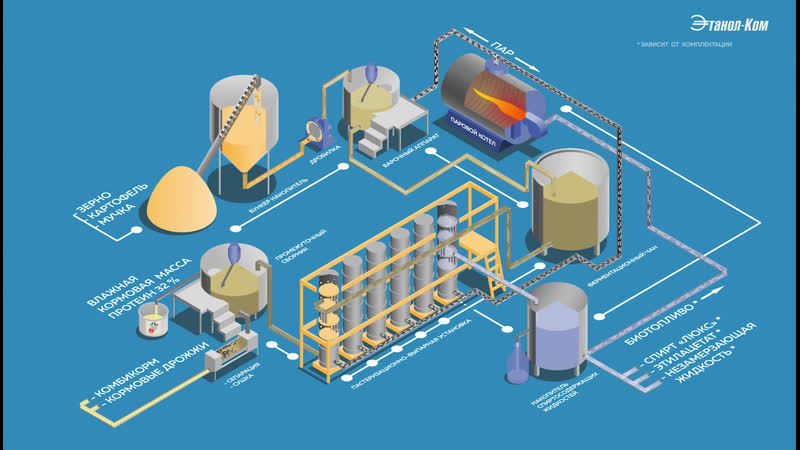 Стекающая вниз бражка постепенно освобождается от спирта, а затем из кубовой царги колонны 18 по трубе 21 переходит в теплообменник 41, где нагревает поступающую в колонну бражку до 60—70е. Дальше бражку нагревают до 105° в колонне острым паром, поступающим по трубе 20. Освобожденная от спирта бражка называется «бардой». По трубе 42 Барда выходит из бардяного теплообменника 41 и направляется в дрожжевой цех для получения из пентоз кормовых дрожжей. Этот процесс в дальнейшем будет подробно рассмотрен.
Стекающая вниз бражка постепенно освобождается от спирта, а затем из кубовой царги колонны 18 по трубе 21 переходит в теплообменник 41, где нагревает поступающую в колонну бражку до 60—70е. Дальше бражку нагревают до 105° в колонне острым паром, поступающим по трубе 20. Освобожденная от спирта бражка называется «бардой». По трубе 42 Барда выходит из бардяного теплообменника 41 и направляется в дрожжевой цех для получения из пентоз кормовых дрожжей. Этот процесс в дальнейшем будет подробно рассмотрен.
Бражная колонна в верхней укрепляющей части заканчивается дефлегматором 19, в котором конденсируются пары ьод — носпиртовой смеси, поступающие с верхней тарелки колонны.
В 1 м3 бражки при температуре 30° растворяется около 1 мъ углекислого газа, образовавшегося при брожении. При нагревании бражки в теплообменнике 41 и острым паром в нижней части бражной колонны растворенная углекислота выделяется и вместе с парами спирта поднимается в укрепляющую часть колонны и далее в дефлегматор 19. Неконденсирующиеся газы отделяются через воздушники, установленные на трубопроводах спиртового конденсата после холодильников. Низкокипящие фракции, состоящие из спирта, альдегидов и эфиров, проходят через дефлегматор 19 и окончательно конденсируются в холодильнике 39у Откуда в виде флегмы стекают обратно в колонну через гидрозатвор 40. Неконденсирующиеся газы, состоящие из углекислого газа, перед выходом из холодильника 39 проходят дополнительный конденсатор или промываются в скруббере водой для улавливания последних остатков спиртовых паров.
Неконденсирующиеся газы отделяются через воздушники, установленные на трубопроводах спиртового конденсата после холодильников. Низкокипящие фракции, состоящие из спирта, альдегидов и эфиров, проходят через дефлегматор 19 и окончательно конденсируются в холодильнике 39у Откуда в виде флегмы стекают обратно в колонну через гидрозатвор 40. Неконденсирующиеся газы, состоящие из углекислого газа, перед выходом из холодильника 39 проходят дополнительный конденсатор или промываются в скруббере водой для улавливания последних остатков спиртовых паров.
На верхних тарелках бражной колонны в жидкой фазе содержится 20—40% спирта.
Конденсат по трубе 25 поступает на питающую тарелку ректификационной колонны 22. Эта колонна работает аналогично бражной колонне, но на более высоких концентрациях спирта. В нижнюю часть этой колонны по трубе 24 подается острый пар, который постепенно вываривает спирт из спиртового конденсата, стекающего в низ колонны. Освобожденная от спирта жидкость, называемая лютером, по трубе 23 уходит в канализацию. Содержание спирта в барде и лютере составляет не более 0,02%.
Освобожденная от спирта жидкость, называемая лютером, по трубе 23 уходит в канализацию. Содержание спирта в барде и лютере составляет не более 0,02%.
Над верхней тарелкой ректификационной колонны устанавливается дефлегматор 26. Не сконденсировавшиеся в нем пары окончательно конденсируются в конденсаторе 26а и стекают обратно в колонну. Часть низкокипящих фракций отбирается по трубе 43 в виде эфироальдегидной фракции, которая возвращается в бродильные чаны, если она не имеет применения.
Для освобождения этилового спирта от летучих органических кислот в колонну подается из бака 45 10%-ный раствор едкого натра, который нейтрализует кислоты на средних тарелках укрепляющей части колонны. В средней части ректификационной колонны, где крепость спирта составляет 45—50%, накапливаются сивушные масла, которые отбираются по трубе 46. Сивушные масла представляют собой смесь высших спиртов (бутиловый, пропиловый, амиловый), образовавшихся из аминокислот.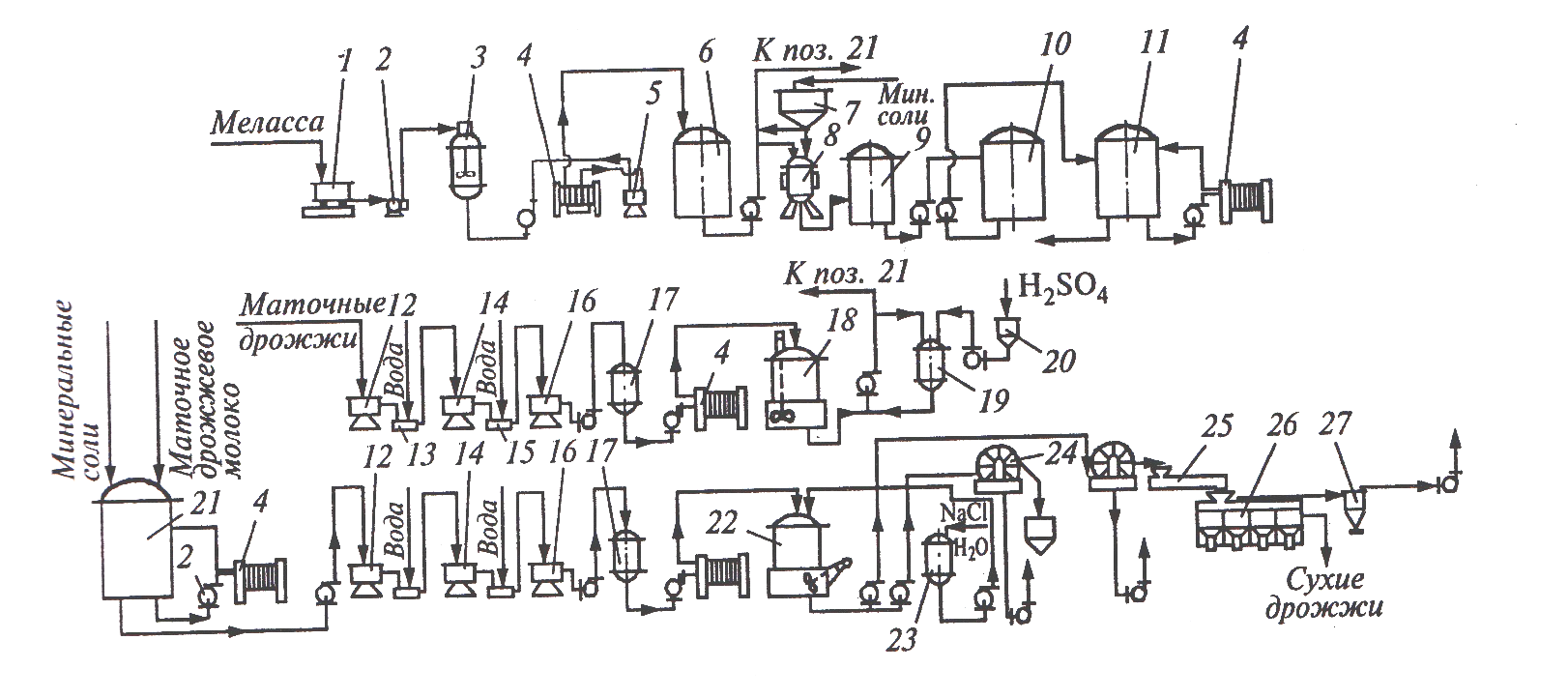
Этиловый спирт, освобожденный от эфиров и альдегидов, а также сивушных масел, отбирается при помощи гребенки с верхних тарелок укрепляющей части ректификационной колонны и по трубе 27 поступает на питающую тарелку метанольной колонны 28. Спирт-сырец, поступающий из ректификационной колонны, содержит около 0,7% метилового спирта, который образовался при гидролизе растительного сырья и вместе с моносахаридами попал в древесное сусло.
При брожении гексоз метиловый спирт не образуется. По техническим условиям на этиловый спирт, вырабатываемый гидролизными заводами, в нем должно содержаться не более 0,1% метилового спирта. Исследования показали, что легче всего метиловый спирт отделяется из спирта-сырца при минимальном содержании в нем воды. По этой причине в метанольную колонну подают спирт-сырец с максимальной крепостью (94—96% этанола). Выше 96%’ этиловый спирт получить на обычных ректификационных колоннах нельзя, так как этой концентрации отвечает состав нераздельнокипящей водоспиртовой смеси.
В метанольной колонне легкокипящей фракцией является метанол, который поднимается в верхнюю часть колонны, укрепляется в дефлегматоре 29 и по трубе 30 сливается в сборники метанольной фракции, содержащей около 80% метанола. Для выпуска товарного 100%-ного метанола устанавливается вторая метанольная колонна, не показанная на рис. 77.
Этиловый спирт, стекая по тарелкам, опускается в нижнюю часть метанольной колонны 28 и по трубе 33 сливается в приемники готовой продукции[2]. Обогревают метанольную колонну глухим паром в выносном подогревателе 31, который установлен таким образом, что по принципу сообщающихся сосудов его межтрубное пространство залито спиртом. Поступающий в подогреватель водяной пар нагревает спирт до кипения и образующиеся спиртовые пары идут на обогрев колонны. Пар, поступающий в подогреватель 31, конденсируется в нем и в виде конденсата подается в сборники чистой воды или сливается в канализацию.
Количество и крепость полученного этилового спирта измеряют в специальной аппаратуре (фонарь, контрольный снаряд, мерник спирта). Из мерника паровым насосом этиловый спирт подают за пределы главного корпуса — в стационарные цистерны, располженные в складе спирта. Из этих цистерн по мере необходимости товарный этиловый спирт переливают в железнодорожные цистерны, в которых отвозят его к местам потребления.
Из мерника паровым насосом этиловый спирт подают за пределы главного корпуса — в стационарные цистерны, располженные в складе спирта. Из этих цистерн по мере необходимости товарный этиловый спирт переливают в железнодорожные цистерны, в которых отвозят его к местам потребления.
Описанный выше технологический процесс дает возможность получать из 1 т абсолютно сухой хвойной древесины 150—180 л 100%-ного этилового спирта. При этом на 1 дкл спирта расхо
Дуется:
Абсолютно сухой древесины в кг. . . . . 55—66;
TOC o «1-3» h z серной кислоты — моаоидрата в кг … . 4,5;
Извести негашеной, 85%-ной в кг…………………………………………………. 4,3;
Пара технологического 3- и 16-атмосферного
В мегакалориях. ………………………………………………………………………….. 0,17—0,26;
Воды в м3……………………………………………………………………………………………. 3,6;
Элекгрознер в квт-ч.…………………………………………………………………….. 4,18
Годовая производительность гидролизно-спиртового завода средней мощности по спирту составляет 1 —1,5 млн.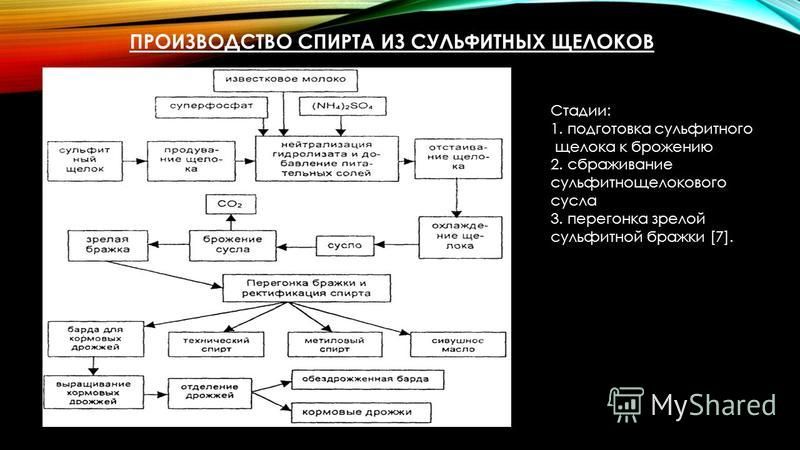 дал. На этих заводах основным продуктом является этиловый спирт. Как уже указывалось, одновременно с ним из отходов основного производства на гидролизно-спиртовом заводе вырабатывается твердая или жидкая углекислота, фурфурол, кормовые дрожжи, продукты переработки лигнина. Эти производства будут рассмотрены в дальнейшем.
дал. На этих заводах основным продуктом является этиловый спирт. Как уже указывалось, одновременно с ним из отходов основного производства на гидролизно-спиртовом заводе вырабатывается твердая или жидкая углекислота, фурфурол, кормовые дрожжи, продукты переработки лигнина. Эти производства будут рассмотрены в дальнейшем.
На некоторых гидролизных заводах, получающих в качестве основного продукта фурфурол или ксилит, после гидролиза богатых пентозами гемицеллюлоз остается трудногидролизуемый остаток, состоящий из целлюлозы и лигнина и носящий название целлолигнина.
Целлолигнин может быть гидролизован перколяционным методом, как описано выше, и полученный гексозный гидролизат, обычно содержащий 2—2,5% Сахаров, может быть переработан по описанной выше методике в технический этиловый спирт или кормовые дрожжи. По этой схеме перерабатывается хлопковая шелуха, кукурузная кочерыжка, дубовая одубина, подсолнечная лузга и т. д. Такой производственный процесс является экономически выгодным только при дешевом сырье и топливе.
На гидролизно-спиртовых заводах обычно получается технический этиловый спирт, используемый для последующей химической переработки. Однако в случае необходимости этот спирт
сравнительно легко очищается путем дополнительной ректификации и окисления щелочным раствором перманганата. После такой очистки этиловый спирт вполне пригоден для пищевых целей.
Комментирование и размещение ссылок запрещено.
Производство — гидролизный спирт — Большая Энциклопедия Нефти и Газа, статья, страница 1
Производство — гидролизный спирт
Cтраница 1
Производство гидролизного спирта из сельскохозяйственных отходов на южных заводах предполагается прекратить из-за его высокой стоимости. [2]
Является побочным продуктом при производстве гидролизного спирта. [3]
Советские ученые и инженеры успешно развивают и рационализируют технологический процесс производства гидролизного спирта, применяя новые прогрессивные методы производства. В 1946 г. группе ученых и инженеров ( Шарков, Чепиго, Мартыненко, Иванов) за разработку и внедрение методов производства этилового спирта из древесины присуждена Сталинская премия.
[4]
В 1946 г. группе ученых и инженеров ( Шарков, Чепиго, Мартыненко, Иванов) за разработку и внедрение методов производства этилового спирта из древесины присуждена Сталинская премия.
[4]
Известны два вида технического лигнина: гидролизный лигнин, получаемый при производстве гидролизного спирта из древесины, и лигнин из сульфитных щелоков — отхода производства целлюлозы сульфитным способом. Химический состав лигнина не установлен, по-видимому, в основе его лежат производные ароматического ряда. В настоящее время установлена возможность частичной ( до 15 %) замены древесной муки лигнином в пресс-композициях темных цветов. [5]
За годы войны ( 1941 — 1945 гг.) в Советском Союзе благодаря развитию производства гидролизного спирта было сэкономлено 10 млн. т зерна; с начала 60 — х годов, благодаря производству этилового спирта из этилена, в год экономится не менее 2 млн.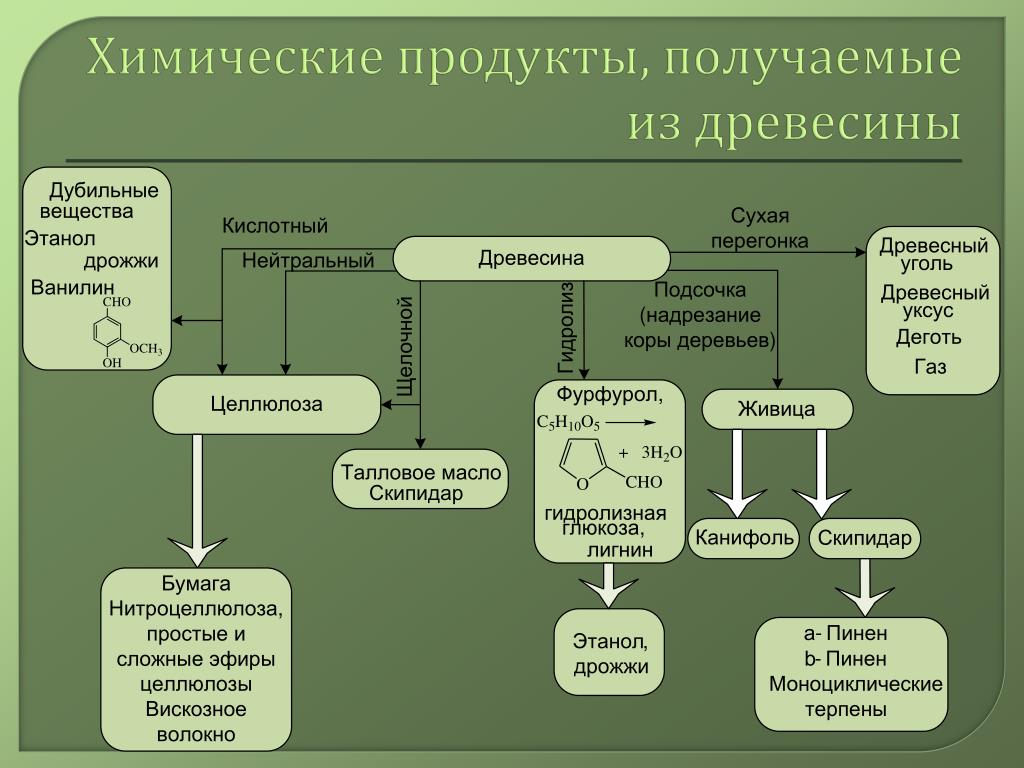 т зерна.
[6]
т зерна.
[6]
Промышленное использование ксилана организуется как самостоятельное производство для переработки растительного сырья67 72 или комбинируется с производством гидролизного спирта и целлюлозы. В последнем случае растительный материал подвергается предварительному гидролизу для удаления пентозанов с последующей их переработкой на фурфурол или ксилозу, а затем производится гидролиз целлюлозы до глюкозы с дальнейшей переработкой в спирт. [7]
Окисление лигнинсодержащего сырья ( например, лигносульфатов — отхода в производстве древесной целлюлозы сульфитной варки, барды — отхода в производстве гидролизного спирта или дрожжей, древесных опилок и др.) в щелочной среде в присутствии оксидов Со, Си, Мп или других катализаторов; из смеси продуктов реакции ванилин экстрагируют бутанолом или бензолом и очищают гидросульфитной обработкой, перегонкой или кристаллизацией. [8]
[8]
Получается из подсолнечной или овсяной лузги, из кендыря, при действии серной кислоты под давлением, и как побочный продукт при производстве гидролизного спирта из древесины. [9]
Указанные обстоятельства определяют целесообразность развития гидролизного производства в Советском Союзе. Производство гидролизного спирта было организовано в Советском Союзе в годы довоенных пятилеток. В настоящее время по производству гидролизного спирта Советский Союз занимает одно из первых мест в мире. [10]
Однако на практике встречаются и обратные задачи. При производстве гидролизного спирта получаются отходы, состоящие из пропитанной раствором серной кислоты клетчатки. При сжигании этих отходов вначале имеет место разложение серной кислоты на HjO я SO3, а затем диссоциация SOs. [11]
[11]
Накапливается как отход производства гидролизного спирта и дрожжей из древесины и другого растительного сырья. [12]
Сульфитный щелок после соответствующей подготовки ( отдувка SO2, нейтрализация, отделение гипсового шлама) поступает в бродильное отделение. Сброженное сусло, так же как и при производстве гидролизного спирта, проходит брагоректификационную колонну, затем спирт укрепляется, чистится от примесей и направляется на склад готовой продукции. [13]
Вначале пищевое сырье в производстве спирта стала вытеснять древесина. Из 1 т древесины при гидролизе получается около 160 кг этилового спирта, что заменяет 1 6 т картофеля или 0 6 т зерна. Производство гидролизного спирта обходится дешевле, чем из пищевого сырья. При комплексной химической переработке древесина используется вместо пищевого сырья также в производстве глицерина, кормового сахара, кормовых дрожжей, уксусной, лимонной и молочной кислот и других продуктов. [14]
[14]
Указанные обстоятельства определяют целесообразность развития гидролизного производства в Советском Союзе. Производство гидролизного спирта было организовано в Советском Союзе в годы довоенных пятилеток. В настоящее время по производству гидролизного спирта Советский Союз занимает одно из первых мест в мире. [15]
Страницы: 1 2
Гидролизное производство — это… Что такое Гидролизное производство?
| Проверить информацию. Необходимо проверить точность фактов и достоверность сведений, изложенных в этой статье. |
В связи с сокращением запасов ископаемого органического сырья в последние годы во всём мире уделяется серьёзное внимание вопросам химической и биотехнологической переработки биомассы растительного сырья (фитомассы) — древесины и сельскохозяйственных растений. В отличие от ископаемых источников органического сырья запасы фитомассы возобновляются в результате деятельности высших растений. Ежегодно на нашей планете образуется около 200 млрд. т растительной целлюлозосодержащей биомассы. Биосинтез целлюлозы — самый крупномасштабный синтез в прошлом, настоящем и будущем.
В отличие от ископаемых источников органического сырья запасы фитомассы возобновляются в результате деятельности высших растений. Ежегодно на нашей планете образуется около 200 млрд. т растительной целлюлозосодержащей биомассы. Биосинтез целлюлозы — самый крупномасштабный синтез в прошлом, настоящем и будущем.
В настоящем столетии основными видами органического сырья будут древесина, сельскохозяйственные растения и каменный уголь, в свою очередь имеющий растительное происхождение.
По ориентировочным оценкам мировые разведанные запасы нефти примерно равны запасам древесины на нашей планете, однако ресурсы нефти быстро истощаются, в то время как в результате естественного прироста запасы древесины могут возобновляться. Значительным резервом повышения ресурсов древесного сырья является увеличение выхода целевых продуктов из древесины. В недалёком будущем нас ожидает переход от нефтехимического производства к биохимической и химической переработке древесины и других видов растительного сырья. Переработка биомассы растительного сырья базируется в основном на сочетании химических и биохимических процессов.
Переработка биомассы растительного сырья базируется в основном на сочетании химических и биохимических процессов.
Под биотехнологией обычно понимают промышленный биохимический синтез ценных веществ и переработку продуктов биологического происхождения. Производственной основой современной биотехнологии является микробиологическая промышленность, включающая гидролизные производства. Эти производства основаны на реакции гидролитического расщепления гликозидных связей полисахаридов биомассы одревесневшего растительного сырья с образованием в качестве основных продуктов реакции моносахаридов, которые подвергаются дальнейшей биохимической или химической переработке, либо входят в состав товарной продукции.
Гидролиз растительного сырья — наиболее перспективный метод химической переработки древесины, так как в сочетании с биотехнологическими процессами позволяет получать кормовые и пищевые продукты, биологически активные препараты, мономеры и синтетические смолы, топливо для двигателей внутреннего сгорания и разнообразные продукты для технических целей.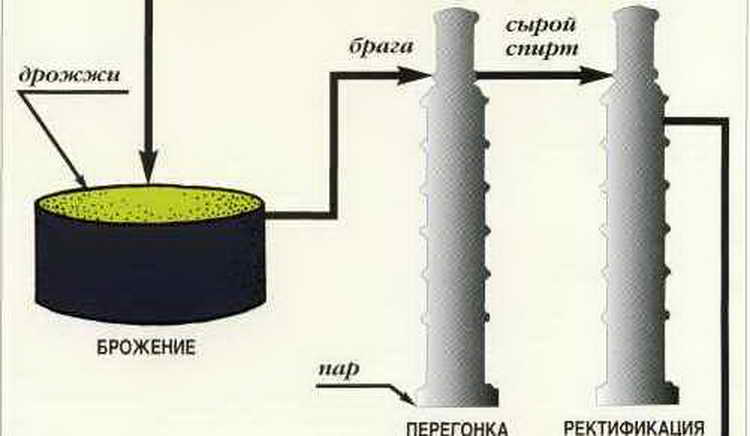
Основные этапы развития гидролизных производств
Созданию отечественной гидролизной промышленности предшествовали многолетние исследовательские и опытные работы, которые обеспечили необходимые предпосылки для разработки современной технологии гидролиза растительного сырья и получения этанола, кормовых дрожжей, фурфурола, ксилита и других продуктов.
В 1811 году впервые систематические исследования по гидролизу растительного полисахарида — крахмала провёл в Петербурге Константин Кирхгоф. При гидролизе крахмала 0,5%-ным раствором C2H5OH при кипячении в течение 24—25 ч из 100 кг крахмала им было получено 75 кг кристаллической глюкозы.
В 1819 году французский химик Браконно получил глюкозу при гидролизе льняного полотна и древесины 91,5%-ной серной кислотой.
В 1822 году русский химик П. Фогель для гидролиза древесины и других целлюлозных материалов предложил применять 87%-ную H2SO4, которая не вызывала обугливания части вещества.
В 1832 году — первая публикация немецкого химика Доберейнера о случайном открытии фурфурола при попытке получения муравьиной кислоты путём обработки сахара и крахмала с помощью H2SO4, а в 1840 г. фурфурол был впервые получен Стейнхаузом из древесных опилок и соломы.
1844 год — первые опыты по гидролизу древесины разбавленной серной кислотой в автоклаве под повышенным давлением, проведённые Пайеном.
1854 год — Пелуз и Арну во Франции разработали метод получения винного спирта из древесины путём её гидролиза концентрированной серной кислотой при комнатной температуре. Этот способ был положен в основу технологии на гидролизной установке, построенной в Париже в 1855 г. В том же году в Брюсселе Мелсеном была разработана технология получения глюкозы и этанола гидролизом измельчённой древесины 2—5%-ной H2SO4 при 100—170 °C.
В 1877 году русский химик М. Чирвинский провёл гидролиз древесных опилок 0,5—1%-ным и растворами H2SO4 и HCl и рекомендовал использовать получаемый сахар для кормовых целей.
В 1891 году Бертраном и Фишером впервые был синтезирован ксилит путём восстановления D— и L—ксилозы амальгамой натрия.
В 1894 году шведским химиком Симонсеном предложен следующий режим одноступенчатого гидролиза: продолжительность гидролиза 25 мин при концентрации H2SO4 0,5 % и температуре 165—170 °C, жидкостной модуль 5, выход сахара 22—23 % от массы древесины, расход H2SO4 2,5—3 %. На полузаводской установке по этому методу выход спирта составлял 50—60 л из 1 т абс. сухой древесины.
В 1899 году на опытной установке в г. Архангельске под руководством О. Гиллер—Бомбина впервые был испытан батарейный метод гидролиза с последовательным пропусканием гидролизата через ряд автоклавов, в результате чего концентрация сахара повышалась до 12—14 %.
В 1899 году профессор Кляссен в Германии предложил проводить гидролиз древесины сернистой кислотой. По этой технологии были построены гидролизно-спиртовые заводы во Франции, Англии и США, которые вскоре были закрыты из-за технических неполадок.
В 1910 году в г. Джорджтаун (США) по методу Симонсена был пущен в эксплуатацию гидролизно-спиртовой завод. Выход спирта на этом заводе достигал 53—73 л из 1 т сухих опилок. В 1913 году по той же технологии был пущен завод в г. Фуллертоне с суточной производственной мощностью 1 000 дал спирта. Заводы работали в течение 5—8 лет.
В 1919 году в Петрограде М. М. Шмидтом на Охтенском заводе организовано получение глюкозной патоки и кристаллической глюкозы из древесных опилок и целлюлозы путём гидролиза концентрированной H2SO4 с последующей регенерацией кислоты методом диализа через растительный пергамент.
В 1922 г. начато освоение промышленной технологии фурфурола в США. Майнер и Браунли на опытно-промышленной установке получили первые тонны фурфурола при переработке овсяной шелухи.
В 1926 году немецким химиком Бергиусом при участии Хегглунда (Швеция) была разработана технология гидролиза древесины концентрированной соляной кислотой. Технология проверена на опытной установке в Маннгейме—Рейнау, где в 1933 г.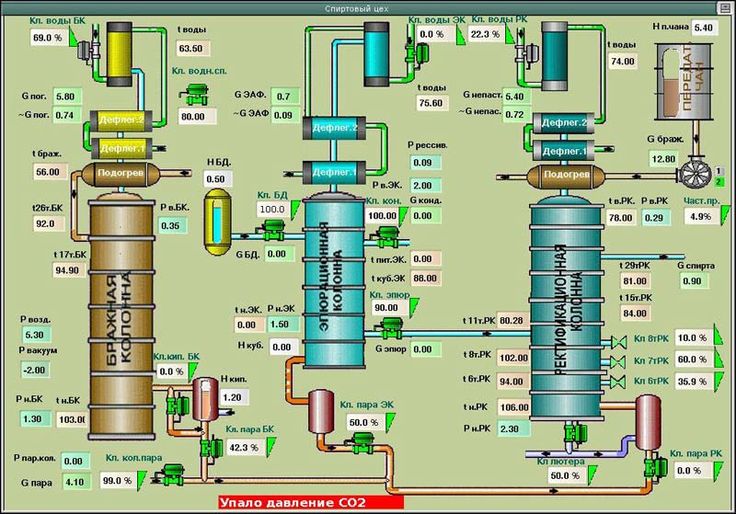 был пущен завод мощностью 6—8 тыс. т кормового сахара в год. С 1934 г. налажено производство кристаллической глюкозы и этанола, с 1939 г. вместо этанола получали пищевые дрожжи. В 1941 г. пущен завод пищевых дрожжей в г. Регенсбурге по технологии Бергиуса.
был пущен завод мощностью 6—8 тыс. т кормового сахара в год. С 1934 г. налажено производство кристаллической глюкозы и этанола, с 1939 г. вместо этанола получали пищевые дрожжи. В 1941 г. пущен завод пищевых дрожжей в г. Регенсбурге по технологии Бергиуса.
В 1926 г. немецкий химик Шоллер предложил принцип перколяционного гидролиза, предусматривающий выведение преобразовавшегося сахара из реакционного объёма током перколирующей кислоты. В 1931 году был пущен первый гидролизный завод в г. Торнеше, где проводился многоступенчатый гидролиз древесины (прерывистая перколяция). К 1934 году на этом заводе было получено 6 000 т сахара, из которого выработано 2,5 млн л спирта. В 1936 году пущен завод по технологии Шоллера в г. Дессау и в 1937 году — в г. Хольцминдене общей мощностью 0,85 млн дал спирта в год.
В 1930 г. было организовано производство фурфурола в СССР на установках малой мощности.
Таким образом, к началу 1930—х годов имелись результаты экспериментальных исследований, опытных работ и определённый практический опыт промышленной реализации гидролизной технологии с получением этанола и фурфурола..JPG)
В СССР в это время по методу акад. С. В. Лебедева интенсивно развивалось производство синтетического каучука с использованием в качестве сырья этанола. Перед научными коллективами страны была поставлена задача в кратчайшие сроки разработать и реализовать в промышленных условиях технологию получения технического этанола из непищевых видов сырья, в частности из древесины. К решению этой проблемы был привлечён Центральный научно-исследовательский лесохимический институт (ЦНИЛХИ) в г. Москве, где проводились работы по гидролизу древесины концентрированной серной кислотой под руководством проф. Л. П. Жеребова. Гидролиз разбавленными кислотами исследовали в Ленинградской лесотехнической академии под руководством В. И. Шаркова, в Ленинградском научно-исследовательском лесохимическом институте (ЛенНИИЛХИ) под руководством Н. Я. Солечника, а также в г. Архангельске и Верхнеднепровске, где позже были организованы опытные производства спирта путём гидролиза древесины и растительных сельскохозяйственных отходов.
Задача создания отечественной технологии получения этанола из древесины была успешно решена в лаборатории гидролиза древесины Ленинградской лесотехнической академии. Осенью 1931 г. В. И. Шарковым, О. Д. Камалдиной, Т. В. Утцаль были проведены первые опыты по гидролизу древесных опилок разбавленной серной кислотой. В начале 1932 г. в состав исследовательской группы вошли И. А. Беляевский, В. В. Головин, А. И. Скриган, А. П. Петроченко и др. За очень короткий период времени была разработана технология гидролиза древесины разбавленной серной кислотой с получением этанола и начато проектирование опытного Череповецкого завода. При разработке оборудования этого завода принимали участие проф. Н. Н. Непенин, Г. М. Орлов и другие специалисты.
В январе 1934 г. был пущен Череповецкий опытный завод, оборудованный 6 гидролизаппаратами вместимостью 1 м³. В конце года выход спирта составил 178 л из 1 т абс. сухого сырья. За 5 лет работы этого завода были проверены многочисленные технологические режимы, на основе которых выбраны параметры гидролиза, подготовки и биохимической переработки гидролизата, ректификационного концентрирования и очистки этанола.
Наряду с проведением исследовательских, проектных и опытных работ под руководством В. И. Шаркова в Ленинградской лесотехнической академии в это же время по специальной программе готовились кадры для гидролизной промышленности. В 1934 г. была организована первая в стране кафедра гидролизных производств. Первый выпуск инженеров по гидролизной специальности состоялся в 1935 г. В 1936 г. было опубликовано первое учебное пособие для вузов В. И. Шаркова «Гидролиз древесины». До последнего дня жизни профессор В. И. Шарков руководил кафедрой гидролизных производств лесотехнической академии, ректором которой он был в период с 1964 по 1973 гг. Выпускники академии стали основным творческим ядром проектного института Гипрогидролиз, организованного в 1934 г. для проектирования гидролизных заводов, и Всесоюзного научно-исследовательского института гидролизной и сульфитно-спиртовой промышленности (ВНИИГС), образованного в 1939 г.
В 1934 г. было принято решение о строительстве первых гидролизных заводов — Ленинградского, Бобруйского, Хорского, Архангельского, Саратовского и Волгоградского. В середине 1934 г. было начато строительство Ленинградского гидролизного завода и в декабре 1935 г. завод был пущен в эксплуатацию. Большую помощь при строительстве этого завода оказала ленинградская партийная организация и лично С. М. Киров, который посещал строительство и не раз оказывал помощь при решении организационных вопросов. Технология гидролиза и получения спирта после пуска завода была несовершенной. Гидролиз опилок проводили 0,5—0,7%-ным раствором H2SO4 при 175—190 °C в стальных футерованных гидролизаппаратах объёмом 18 м³. При пуске завода использовали одноступенчатый (так называемый стационарный) метод гидролиза, к 1941 г. был освоен оросительный вариант перколяционного гидролиза, а в 1945 г. был разработан режим перколяционного гидролиза с запасом жидкости, соответствующим уровню сырья (ленинградский режим гидролиза).
В 1936 и 1939 гг. введены в эксплуатацию Бобруйский и Хорский гидролизные заводы. В 1939 г. организовано Главное управление гидролизной и сульфитно-спиртовой промышленности при Совнаркоме СССР. Таким образом, создание гидролизной промышленности проходило в годы первых пятилеток, в период индустриализации народного хозяйства страны.
В годы Великой Отечественной войны потребность в техническом этаноле резко возросла, так как он применялся для оборонных целей. В период с 1941 по 1945 гг. введены в эксплуатацию Архангельский, Саратовский, Канский, Красноярский, Тавдинский и Волгоградский заводы. В 1943 г. пущен первый цех по получению кормовых дрожжей на Саратовском заводе, в 1944 г. — на Хорском заводе.
Выпуск гидролизной продукции имел большое значение во время блокады г. Ленинграда. В октябре 1941 г. на совещании в Смольном проф. В. И. Шарков предложил организовать производство гидроцеллюлозы («пищевой» целлюлозы) для использования в качестве структурной добавки (10—15 %) к ржаному хлебу и пищевых белковых дрожжей. При участии доцентов кафедры гидролизных производств лесотехнической академии А. В. Буевского и Ф. А. Сартания, научных сотрудников ВНИИГСа М. Я. Калюжного, Н. П. Мельникова, В. И. Тимофеевой и др. производство этих продуктов было организовано на Ленинградском гидролизном заводе при использовании оборудования, оставшегося после эвакуации этого завода. Завод выдавал продукцию с ноября 1941 г. по декабрь 1943 г. Цехи по производству гидроцеллюлозы и пищевых дрожжей были также организованы на других предприятиях города.
Последующие этапы становления и развития отечественной гидролизной промышленности можно представить в виде краткой хронологической сводки.
1946 г. — пуск первого завода по переработке хлопковой шелухи — Ферганского гидролизного завода. В том же году было организовано промышленное производство фурфурола из конденсатов паров самоиспарения гидролизата хвойной древесины на гидролизно-спиртовых заводах — Канском, Красноярском, Тавдинском.
1959—1962 гг. — промышленное освоение пентозного гидролиза хлопковой шелухи с получением ксилитана и триоксиглутаровой кислоты.
1955—1974 гг. — опытное производство глюкозы на Канском гидролизном заводе при гидролизе древесины концентрированной соляной кислотой.
1962—1963 гг. — пуск первых заводов гидролизно-дрожжевого профиля: Аполлонского и Кропоткинского.
1962 г. — ввод в эксплуатацию первого цеха по получению активного угля — коллактивита из технического лигнина.
1960—1968 гг. — организация производства фурфурилового и тетрагидрофурфурилового спиртов, тетрагидрофурана, лаков и смол на основе фурфурола и его производных.
1966 г. — организована микробиологическая промышленность СССР, в состав которой в качестве подотрасли вошла гидролизная промышленность.
1967 г. — пущен ксилитный цех на Ферганском химическом заводе фурановых соединений. 1973—1974 гг. введены в эксплуатацию крупнейшие биохимические заводы дрожжевого профиля — Кировский, Лесозаводский, Волжский, а в 1975 г. — освоено производство дрожжевых премиксов на Бельцком и Кропоткинских заводах.
1977 г. — начато получение фурфурола из лиственной древесины на заводах фурфурольно-дрожжевого профиля.
1979 г. — освоено производство гидролизного кормового сахара на Кировском заводе.
1981 г. — в СССР выработано 1 млн т кормовых дрожжей (на углеводном и углеводородном сырье).
1982 г. — на Речицком гидролизно-дрожжевом заводе в опытно-промышленных масштабах освоено производство растительно-углеводных кормовых добавок.
1985 г. — гидролизные заводы вошли в состав Министерства медицинской и микробиологической промышленности СССР.
Современное состояние гидролизных производств
Перспективы развития гидролитической и биотехнологической переработки растительного сырья
Проекты заводов этанола из биомассы (гидролизные заводы)
В 2007 году DOE выделил финансирование трёх проектов в размере $160 млн., но после ежегодного послания президента США, финансирование было увеличено. В своём послании Конгрессу в январе 2007 г. Дж.Буш призвал сократить потребление бензина на 20% за 10 лет (план «20 за 10»). Всего инвестиции в шесть биоэнергетических проектов составят $1,2 млрд. Эти заводы ежегодно будут производить из целлюлозы 130 млн. галлонов (490 млн. литров) этанола. Инвестиции будут сделаны:
Abengoa Bioenergy Biomass (Канзас): Грант — $76 млн. Мощность завода 44 млн. литров этанола в год. Ежедневно будет перерабатываться 700 тонн стеблей кукурузы, пшеничной соломы, и др. отходов.
ALICO, Inc. (Флорида): Грант — $33 млн. Мощность завода 54 млн. литров этанола в год, а также ежедневно электроэнергия, 8.8 тонн водорода и 50 тонн аммиака. Ежедневно будет перерабатываться 770 тонн лесных отходов и отходов сахарного тростника.
BlueFire Ethanol (Калифорния): Грант — $40 млн. Мощность — 74 млн. литров этанола в год из 700 тонн в день древесных и растительных отходов.
Broin Companies (Южная Дакота): Грант — $80 млн. Мощность — 484 млн. литров этанола в год из них 25% из целлюлозы. Ежедневно будет перерабатываться 842 тонны соломы и отходов кукурузы.
Iogen Biorefinery Partners (Виргиния): Грант — $80 млн. Мощность — 70 млн. литров этанола в год. Ежедневно будет перерабатываться 700 тонн соломы пшеницы, ячменя, риса, кукурузы.
Range Fuels (Колорадо): Грант — $76 млн. Мощность — 160 млн. литров этанола в год и 36 млн. литров метанола. Ежедневно будет перерабатываться 1200 тонн древесных отходов.
См. также
Ссылки
Спирт из опилок в домашних условиях видео – АвтоТоп
Вы в лесу. Вокруг теснятся толстые и тонкие стволы деревьев. Для химика все они состоят из одного и того же материала — древесины, основной частью которой является органическое вещество — клетчатка (C6H10O5) х. Клетчатка образует стенки клеток растений, т. е. их механический скелет; довольно чистую мы её имеем в волокнах хлопчатой бумаги и льна; в деревьях она встречается всегда вместе с другими веществами, чаще всего с лигнином, почти такого же химического состава, но обладающего иными свойствами. Элементарная формула клетчатки C6H10O5 совпадает с формулой крахмала, свекловичный сахар имеет формулу C12H22O11. Отношение числа атомов водорода к числу атомов кислорода в этих формулах такое же, как и в воде: 2:1. Поэтому эти и им подобные вещества в 1844 г. были названы «углеводами», т. е. веществами, как бы (но не на самом деле) состоящими из углерода и воды.
Углевод клетчатка имеет большой молекулярный вес. Молекулы её представляют длинные цепи, составленные из отдельных звеньев. В отличие от белых зёрен крахмала, клетчатка представляет прочные нити и волокна. Это объясняется различным, теперь точно установленным, структурным строением молекул крахмала и клетчатки. Чистая клетчатка в технике зовётся целлюлозой.
В 1811 г. академик Кирхгоф сделал важное открытие. Он взял обыкновенный крахмал, полученный из картофеля, и подействовал на него разбавленной серной кислотой. Под действием H2SO4 произошёл гидролиз крахмала и он превратился в сахар:
Эта реакция имела важное практическое значение. На ней основано крахмало-паточное производство.
Но ведь клетчатка имеет ту же самую эмпирическую формулу, что и крахмал! Значит, из неё тоже можно получить сахар.
Действительно, в 1819 г. было впервые осуществлено и осахаривание клетчатки с помощью разбавленной серной кислоты. Для этих целей можно применять и концентрированную кислоту; русский химик Фогель в 1822 г. получил сахар из обычной бумаги, действуя на неё 87-процентным раствором H2SO4.
В конце XIX в. получение сахара и спирта из дерева стало интересовать уже и инженеров-практиков. В настоящее время спирт из целлюлозы получают в заводских масштабах. Способ, открытый в пробирке учёного, стад осуществляться в больших стальных аппаратах инженера.
Посетим гидролизный завод. В огромные варочные котлы (перколяторы) загружают опилки, стружки или щепу. Это — отходы лесопильных или деревообрабатывающих предприятий. Раньше эти ценные отходы сжигались или просто выбрасывались на свалку. Через перколяторы непрерывным током проходит слабый (0,2—0,6%) раствор минеральной кислоты (чаще всего серной). Долго держать одну и ту же кислоту в аппарате нельзя: содержащийся в ней сахар, полученный из древесины, легко разрушается. В перколяторах давление 8—10 ат, а температура 170—185°. При этих условиях гидролиз целлюлозы идёт значительно лучше, чем при обычных условиях, когда процесс весьма затруднителен. Из перколяторов получают раствор, содержащий около 4% сахара. Выход сахаристых веществ при гидролизе достигает 85 % от теоретически возможного (по уравнению реакции).
Дальше сахарный раствор поступает на сбраживание в спирт с помощью дрожжей, с чем мы уже знакомы. Так из дерева получают этиловый спирт. Он называется гидролизным спиртом.
Рис. 8. Наглядная схема получения гидролизного спирта из древесины.
Для Советского Союза, имеющего необозримые лесные массивы и неуклонно развивающего промышленность синтетического каучука, получение спирта из древесины представляет особый интерес. Ещё в 1934 г. XVII съезд ВКП(б) постановил всемерно развивать производство спирта из опилок и отходов бумажной промышленности. Первые советские гидролизно-спиртовые заводы начали регулярно работать с 1938 г. За годы второй и третьей пятилеток у нас были построены и пущены заводы по выработке гидролизного спирта — спирта из древесины. Этот спирт в настоящее время всё в больших количествах перерабатывается в синтетический каучук. Это — спирт из непищевого сырья. Каждый миллион литров гидролизного этилового спирта освобождает для питания около 3 тыс. тонн хлеба или 10 тыс. тонн картофеля и, следовательно, около 600 га посевной площади. Для получения этого количества гидролизного спирта нужно 10 тыс. тонн опилок с 45-процентной влажностью, что может дать за год работы один лесопильный завод средней производительности.
На сегодняшний день достаточно много людей занимается изготовлением домашней наливки, однако для некоторых напитков необходимо наличие спиртного элемента. Производство спирта в домашних условиях не является сильно трудоемким. Для этого необходимо знать и учитывать некоторые аспекты и принципы изготовления метилового спирта.
В первую очередь для изготовления метанола требуется наличие зерна. В роли зерновых культур в данном случае могут выступать кукуруза, пшеница. Также можно использовать картофель и крахмал. Но, как известно, во взаимодействии с веществом крахмал не дает никакой реакции. С целью произвести химический элемент используется метод засахарения. А для того, чтобы его засахарить, необходимы определенные ферменты, они присутствуют в солоде. Делая этанол из зерна без химических примесей, наблюдается выход натурального продукта.
Технология производства метанола
Технология по производству спиртового химического вещества в домашних условиях может состоять из нескольких этапов.
Ниже представляются самые основные:
- Производство метанола с помощью солода. Зерна культурных растений необходимо проращивать в небольших посудинах, при этом их рассыпают в один слой, примерно до трех сантиметров. Помните, что предварительно пророщенные зерна необходимо обработать раствором марганцовки. После обработки семена помещаются в емкость и смачиваются водой. Следует учитывать, что наличие солнечных лучей, или достаточность света напрямую зависит от скорости прорастания зерна. Поверх емкости следует накрыть полиэтиленовый материал или тонкое стекло, то есть он должен быть достаточно прозрачным. Если наблюдается уменьшение количества воды, ее необходимо добавлять.
- Следующий этап: обработка крахмала. Для начала добываем крахмал из продукта, который выбран для изготовления этанола. В данном случае это картофель. Слегка порченый картофель необходимо варить до тех пор, пока из воды начнет образовываться клейстер. Далее ждем, пока продукт остынет, тем временем измельчаем солод. Следом перемешиваем два продукта. Далее происходит процедура расщепления крахмала, ее необходимо производить при температуре не менее 60 ˚ С. Теперь смесь помещается в посуду с горячей водой и оставляется на 1 час. По истечении времени изделие полностью остужают.
- Этап брожения. Как известно, брожение характеризуется присутствием в алкоголе содержащих элементов. Однако брагу назвать алкогольным напитком невозможно. После остывания смеси добавляются дрожжи, которые способны вступить в реакцию даже при комнатной температуре. Однако если температура поднимается выше — брожение продукта естественно будет происходить быстрее. При значительной жаре процедура брожения закончится по истечении трех суток. При этом из продукта можно ощущать мягкий запах зерна.
- Следующий этап — это перегонка. С помощью чего она производится? Для этого используется специальный аппарат для производства спирта в домашних условиях.
- Заключительным этапом считается технология очистки. Можно сказать, что метиловый спирт готов, но замечается, что жидкость не прозрачная. Именно поэтому и делается очистка. Она проводится путем добавления раствора марганцовки. В таком виде оставляем метиловый спирт на одни сутки, затем фильтруем — продукт готов.
Как, видим, технология изготовления домашнего спирта довольно проста и не требует дополнительных усилий.
Производство этанолового вещества из опилок
В последние годы значительно снизилось ископаемое сырье, которое можно использовать для изготовления этилового спирта. Наблюдается нехватка зерна. Однако производство спирта из древесных опилок не самый худший вариант, так как этот сырьевой продукт постоянно обновляется по истечении годов.
Однако изготовление вещества из опилок требует некоторых навыков, и плюс ко всему изготовитель должен иметь специальное оборудование, без которого будет трудоемко изготавливать этанол. Производство спирта из опилок в домашних условиях пользуется высокой популярностью, так не требует высоких затрат.
Как известно, свой изготовленный этанол не сравнивается с заводским вариантом. Продукция, изготовленная в хозяйственных условиях, является более качественной, ведь каждый ингредиент отличается своей уникальностью. Из опилок производить спирт значительно проще!
Как производить спиртной продукт дома?
Производство этилового спирта в домашних условиях ведется при использовании специального аппарата. Данный аппарат способен производить процедуру расщепления определенных элементов, а также проводить химические реакции между ними. Обычное оборудование для изготовления спиртной продукции может выглядеть как мини заводы. Изготавливать в них можно любые виды алкогольных напитков.
Изучить технологию приготовления этилового вещества довольно просто, при этом изделие получается высококачественным. Что из этого можно получить? Во-первых, это продукция алкогольного характера с высоким качеством, а во-вторых полностью происходит окупаемость собственных затрат, для этого требуется специальный аппарат.
Например, если используется сахар в количестве 20 кг, с него выходит до 12 литров алкоголя. При этом процент метанола достигает до 96%. Из этого расчета выходит 25 бутылок водки по пол литра. Кроме того, электричества, которое потребляет аппарат, будет потрачено около 25 квт.
Такое оборудование способно использовать все загруженные продукты по назначению. Выход продукта, непригодного для питья, производимого при первой обработке, можно использовать как очиститель для стеклянных поверхностей и окон. Также такой аппарат можно установить самостоятельно, пользуясь необходимыми схемами и чертежами. Такое оборудование с легкостью справится с производством метилового спирта.
Оборудование по производство спиртных продуктов имеет некоторые принципы своей работы. Аппарат имеет специальную горловину, которая заполняет бак необходимой жидкостью. В виде такой жидкости может выступать брага. При помощи нагревательных горелок продукт нагревается до температуры кипения. После чего аппарат и оборудование необходимо перевести в обычный режим.
Далее происходит охлаждение через холодильное отделение с добавочной очисткой пара от ненужных примесей. Очищенное вещество попадает в бак, а пары в холодильник, в котором охлаждаются до состояния жидкости. Аппарат для производства спирта способен выработать установленный норматив. Результатом проведения данной процедуры выступает алкоголь высококачественного приготовления.
В советское время, кто еще помнит, много шутили на тему спирта, приготовленного из опилок. Ходили слухи, будто после войны дешевую водку делали как раз на основе «опилочного» алкоголя. В народе этот напиток получил название – «сучок».
Вообще, разговоры о производстве спирта из опилок возникли, конечно же, не на пустом месте. Такой продукт действительно производился. Назывался он «гидролизным спиртом». Сырьем для его производства действительно были опилки, точнее – целлюлоза, извлекаемая из отходов лесной промышленности. Выражаясь строго научно – из непищевого растительного сырья. По приблизительным расчетам, из 1 тонны древесины можно было получить около 200 литров этилового спирта. Это будто бы позволяло заменить 1,5 тонны картофеля или 0,7 тонн зерна. Применялся ли такой спирт на советских лекеро-водочных заводах, неизвестно. Производился он, понятное дело, для сугубо технических целей.
Надо сказать, что производство технического этанола из органических отходов уже давно будоражит воображение ученых. Можно найти литературу XIX века, где обсуждаются возможности получения спирта из самого разнообразного сырья, в том числе и непищевого. В XX веке эта тема зазвучала с новой силой. В 1920-го годы ученые в Советской России даже предлагали делать спирт из… фекалий! Было даже шутливое стихотворение Демьяна Бедного:
Ну настали времена,
Что ни день, то чудо:
Водку гонят из говна –
По три литра с пуда!
Русский ум изобретет
В зависть всей Европы –
Скоро водка потечет
В рот из самой жопы…
Впрочем, идея с фекалиями так и осталась на уровне шутки. А вот к целлюлозе отнеслись серьезно. Помните, в «Золотом теленке» Остап Бендер рассказывает иностранцам о рецепте «табуреточного самогона». Дело в том, что с целлюлозой «химичили» уже тогда. Причем, надо заметить, извлекать ее можно не только из отходов лесной промышленности. Отечественное сельское хозяйство ежегодно оставляет огромные горы соломы – это тоже прекрасный источник целлюлозы. Не пропадать же добру. Солома – источник возобновляемый, можно сказать – даровой.
Есть только в этом деле одна загвоздка. Помимо нужной и полезной целлюлозы в одревесневших частях растений (а таковыми, в том числе, является и солома) содержится лигнин, который усложняет весь процесс. Из-за наличия в растворе этого самого лигнина практически невозможно получить нормальную «бражку», поскольку сырье не осахаривается. Лигнин тормозит развитие микроорганизмов. По этой причине требуется «подкормка» – добавление нормального пищевого сырья. Чаще всего в этой роли выступает мука, крахмал или патока.
От лигнина, конечно же, можно избавиться. В целлюлозо-бумажной промышленности это традиционно делается химическим путем, например, с помощью обработки кислотой. Вопрос только в том, куда его потом девать? В принципе, из лигнина можно получить неплохое твердое топливо. Горит он хорошо. Так, в Институте теплофизики СО РАН даже разработали соответствующую технологию сжигания лигнина. Но, к сожалению, тот лигнин, что остается от нашего целлюлозо-бумажного производства, в качестве топлива непригоден из-за содержащейся в нем серы (последствия химической обработки). Если его сжигать – получим кислотные дожди.
Есть и другие способы – обрабатывать сырье перегретым паром (лигнин при высоких температурах плавится), проводить экстракцию органическими растворителями. Кое-где именно так и делают, однако эти способы очень затратные. В условиях плановой экономики, где все затраты брало на себя государство, можно было работать и таким способом. Однако в условиях рыночной экономики получается так, что овчинка, образно говоря, не стоит выделки. И при сопоставлении затрат выходит, что куда дешевле обходится производство технического спирта (по-современному – биоэтанола) из традиционного пищевого сырья. Все зависит от того, в каких количествах вы располагаете таким сырьем. У американцев, например, имеет место перепроизводство кукурузы. Куда проще и выгоднее излишки пустить на производство спирта, чем транспортировать ее на другой континент. В Бразилии, как мы знаем, излишки сахарного тростника также используются в качестве сырья для производства биоэтанола. В принципе, в мире не так уж мало стран, где спирт заливают не только в желудок, но и в бак автомобиля. И все было бы неплохо, если бы некоторые известные мировые деятели (в частности, кубинский лидер Фидель Кастро), не выступили против такого «несправедливого» использования сельхозпродукции в условиях, когда в некоторых странах люди страдают от недоедания, а то и вообще умирают с голоду.
В общем, идя навстречу филантропическим пожеланиям, ученые, работающие в сфере производства биоэтанола, должны искать какие-то более рациональные, более совершенные технологии переработки непищевого сырья. Примерно десять лет назад специалисты Института химии твердого тела и механохимии СО РАН решили пойти другим путем – использовать для этих целей механохимический способ. Вместо известной химической обработки сырья или нагревания они стали применять особую механическую обработку. Для чего были сконструированы специальные мельницы и активаторы. Суть метода такова. Благодаря механической активации целлюлоза переходит из кристаллического состояния в аморфное. Это облегчает работу ферментов. Но главное здесь то, что сырье в процессе механической обработки разделяется на различные частички – с разным (большим или меньшим) содержанием лигнина. Потом уже – благодаря разным аэродинамическим характеристикам этих частичек – их легко отделить друг от друга с помощью специальных установок.
На первый взгляд, все очень просто: размололи – и дело с концом. Но только на первый взгляд. Если бы действительно все было так просто, то уже во всех странах мололи бы солому и прочие растительные отходы. На самом деле здесь необходимо найти правильную интенсивность, чтобы сырье разделилось на отдельные ткани. В противном случае у вас получится однообразная масса. Задача ученых – как раз найти здесь необходимый оптимум. И оптимум этот, как показывает практика, достаточно узкий. Можно и перестараться. В том-то, надо сказать, и заключается работа ученого, чтобы выявить золотую середину. Причем, здесь необходимо учитывать и экономические аспекты – а именно, отработать технологию так, чтобы затраты на механихимическую обработку исходного сырья (каким бы дешевым оно ни было) не сказались на себестоимости производства.
В лабораторных условиях уже получены десятки литров замечательного спирта. Самым впечатляющим выглядит тот момент, что спирт получен из обычной соломы. Причем – без применения кислоты, щелочей и перегретого пара. Главное подспорье тут – «чудо-мельницы», сконструированные спецами Института. В принципе, уже ничто не мешает перейти и к промышленным образцам. Но это уже – другая тема.
Вот он – первый отечественный биоэтанол из соломы! Пока еще в бутылях. Дождемся ли, когда его начнут производить цистернами?
Развитие Отечественной гидролизной промышленности Текст научной статьи по специальности «История и археология»
ния, так как позволяет устранять причины оппортунистического поведения участников строительства и уменьшать трансакционные издержки (в части издержек по защите от оппортунизма).
Примечания
’ Уильямсон О.И. Экономические институты капитализма: Фирмы, рынки, «отношенческая» контрактация. СПб., 1996. С. 109.
2 Там же. С. 130.
УДК 332.1(571.53) А.П. СУХОДОЛОВ
ББК 65.9(2) доктор экономических наук,профессор,г. Иркутск
e-mail: [email protected]
В.А. ХАМАТАЕВ
генеральный директор ОАО «Восточно-Сибирский комбинат биотехнологий»,г. Тулун
e-mail: [email protected]
РАЗВИТИЕ ОТЕЧЕСТВЕННОЙ ГИДРОЛИЗНОЙ ПРОМЫШЛЕННОСТИ
Рассмотрено развитие гидролизной промышленности в СССР с конца 1930-х до начала 2000-х гг. Показана роль данной отрасли в годы Великой Отечественной войны и в послевоенный период.
Ключевые слова: СССР, гидролизная промышленность, этапы развития.
Первые опыты получения глюкозы путем гидролиза древесного сырья с использованием растворов серной кислоты проводились в Германии, Франции, Швеции и России в середине XIX в. В частности, в 1855 г. во Франции была смонтирована опытная установка для получения винного спирта из древесины путем ее гидролиза с применением концентрированной серной кислоты при комнатной температуре. Одновременно в Швеции был предложен способ получения глюкозы и этилового спирта посредством гидролиза измельченной древесины с использованием слабого (2-5%-ного) раствора серной кислоты, но при повышенной (170 °С) температуре. Чуть позже, в 1877 г., в России осуществили гидролиз древесных опилок с применением 1%-ного раствора серной кислоты, а продукты гидролиза (сахароподобные вещества) использовали для дрожжевого брожения и получения спирта.
Выход спирта на первых установках был небольшим — всего 50 л из 1 т сухой древесины. Дальнейшее совершенствование технологии гидролиза позволило в начале XX в. создать промышленные установки для получения этанола путем переработки отходов древесного и сельскохозяйственного сырья. В таких странах, как США, Германия, Франция и Англия, стала формироваться
гидролизная промышленность. В дореволюционной России данная отрасль не получила развития, несмотря на разработки русских ученых и наличие огромной сырьевой базы. Первые отечественные гидролизные заводы были построены в СССР только в 1930-х гг.
Становление гидролизной промышленности в СССР. В начале 1930-х гг. в Архангельске и Верхнеднепровске были созданы первые опытно-промышленные заводы по производству спирта путем гидролиза древесины и растительных сельскохозяйственных отходов. В январе 1934 г. дал продукцию уже более крупный, Череповецкий, опытный гидролизный завод, ставший экспериментальной площадкой для отработки наиболее эффективных технологических режимов гидролиза. На этом предприятии удалось получить выход этилового спирта в объеме 170 л из 1 т сухого древесного сырья, что позволило гидролизному производству успешно конкурировать с производством спирта из пищевого сырья.
В течение 1934 г. началось строительство сразу нескольких гидролизных заводов — Ленинградского, Бобруйского, Хорс-кого (на Дальнем Востоке), Саратовского и Волгоградского, были увеличены мощности Архангельского опытно-промышленного завода. Одновременно в Ленинградской лесотехнической академии приступили к подготов-
© А.П. Суходолов, ВА. Хаматаев, 2009
ке кадров для гидролизной промышленности (в 1934 г. была организована первая в стране кафедра гидролизных производств). Первый выпуск инженеров-гидролизников состоялся уже в 1935 г., а еще через год было издано первое учебное пособие для вузов профессора В.И. Шаркова «Гидролиз древесины». Для проектирования гидролизных заводов был создан Всесоюзный НИИ гидролизной и сульфитно-спиртовой промышленности.
В те годы заводы строились и вводились в эксплуатацию очень быстро. Например, Ленинградский гидролизный завод дал первую продукцию уже через полтора года после начала его строительства (в декабре 1935 г.), Бобруйский — через два года. Для оперативного решения проблем развития отрасли в 1939 г. было создано Главное управление гидролизной и сульфитно-спиртовой промышленности при Совнаркоме СССР.
На начальном этапе развития отрасли (до 1943 г.) единственным видом продукции гидролизных заводов являлся этиловый спирт. Необходимость быстрого наращивания его выпуска из непищевого сырья диктовалась острой нехваткой в стране продовольственного зерна (традиционного сырья для производства спирта). В период индустриализации основная его масса экспортировалась, а на валюту закупалось оборудование для промышленных предприятий. Кроме того, в СССР расширялось производство синтетического каучука, требовавшее в качестве сырья большого количества этанола. Наконец, в районах масштабных лесозаготовок скапливались значительные объемы лесных отходов, которые было целесообразно превращать в ценный продукт.
В 1940 г. все гидролизные заводы страны произвели 366 тыс. дал этилового спирта (табл.).
Годы Великой Отечественной войны. В
этот период потребность в этиловом спирте существенно возросла. Он применялся на предприятиях оборонной промышленности, в санитарных подразделениях и тыловых госпиталях, а также в боевых частях Красной Армии, в том числе как легендарные «наркомовские 100 грамм». В те годы гидролизный спирт повсеместно заменил спирт, производимый из пищевого зерна (зерно шло исключительно на продовольственные нужды).
Динамика производства основных видов продукции гидролизной промышленности в СССР
Год Этиловый спирт, тыс. дал Кормовые дрожжи, тыс. т Фурфурол, тыс. т
1940 366 — —
1945 670 0,1 —
1950 3 201 1,1 0,5
1960 14 273 9,8 4,4
1968 19 409 97,1 17,1
1970 18 033 128,3 19,8
1980 13 857 377,7 35,8
1985 24 300 1 413,0 40,1
1990 15 000 400,0 30,0
Мало кто знает, что гидролизная промышленность помогла выжить осажденному Ленинграду. Во время его блокады профессор В.И. Шарков предложил организовать производство пищевых дрожжей и гидролизной («пищевой») целлюлозы и добавлять их при выпечке ржаного хлеба. Эти продукты с ноября 1941 г. по декабрь 1943 г. производились на Ленинградском гидролизном заводе (на оставшемся после эвакуации оборудовании), а также в гидролизных цехах, специально созданных для этих целей на других предприятиях города. Тем самым скудный блокадный паек был увеличен почти на треть.
О важности отрасли свидетельствует и тот факт, что, несмотря на трудности военного времени, в течение 1941-1945 гг. в стране были построены и введены в эксплуатацию достаточно крупные для того времени Саратовский, Канский, Красноярский, Тавдинский и Волгоградский гидролизные заводы. Кроме того, ввиду острой нехватки продовольственных и кормовых ресурсов на гидролизных заводах стали создавать цехи по производству дрожжей. Первые такие цехи пущены в 1943-1944 гг. на Саратовском и Хорском заводах. К концу войны гидролизные предприятия страны производили уже 670 тыс. дал спирта (рост по сравнению с довоенным уровнем составил 1,7 раза).
Послевоенный период. Уже в 1944 г. на гидролизных заводах началась подготовка к выпуску фурфурола, необходимого для скорейшего послевоенного восстановления промышленности и развития новых отраслей. (К тому времени в США уже было налажено масштабное производство подобной продукции, используемой в том числе и в оборонной промышленности.)
Известия ИГЭА. 2009. № 3 (65)
Сразу же после войны были утверждены обширные планы строительства новых и модернизации существующих гидролизных заводов. В частности, в 1946 г. в Ферганской долине был пущен первый завод по переработке хлопковой шелухи, на котором отрабатывалась технология получения новых видов продукции. Было организовано промышленное производство фурфурола из гидролизата хвойной древесины на Канском, Красноярском и Тавдинском гидролизноспиртовых заводах. Именно в послевоенные годы началось строительство крупных гидролизных заводов в Иркутской области.
По своему качеству полученный из древесных отходов спирт не уступал техническому спирту, вырабатываемому из зерна и картофеля. При этом его производство высвобождало для продовольственных целей значительные объемы остродефицитного в то время зерна, что было очень важно в послевоенный восстановительный период.
1950—1980-е гг. Эти годы стали наиболее продуктивными с точки зрения развития отрасли. Кроме строительства новых современных предприятий большое внимание уделялось техническому переоснащению существующих гидролизных заводов, автоматизации основных технологических процессов. Перечислим лишь некоторые факты, свидетельствующие о динамичном развитии отрасли в данный период: организовано производство глюкозы на Канском заводе; пущены первые предприятия гидролизнодрожжевого профиля — Аполлонский и Кропоткинский заводы; налажено производство активированного угля из лигнина, а также стал осуществляться выпуск фурфурилового и тетрагидрофурфурилового спиртов, лаков и смол на основе фурфурола и его производных. Что касается Иркутской области, то здесь были введены в эксплуатацию, а затем модернизированы Бирюсинский, Тулунский и Зиминский гидролизные заводы.
В середине 1960-х гг. было принято решение о создании в СССР микробиологической промышленности, в состав которой в качестве подотрасли вошла гидролизная промышленность. Вскоре были введены в эксплуатацию крупнейшие биохимические заводы дрожжевого профиля — Кировский, Лесозаводский, Волжский, освоен
выпуск кормовых дрожжей на Бельцком и Кропоткинском гидролизных заводах, на Байкальском ЦБК и Братском ЛПК, организовано производство кормового сахара на Кировском гидролизном заводе. Было также начато получение фурфурола из древесины лиственных пород.
В середине 1980-х гг. все гидролизные заводы вошли в состав Министерства медицинской и микробиологической промышленности СССР. Многие из них кооперировались с лесопильно-деревообрабатывающими, целлюлозно-бумажными и сельскохозяйственными предприятиями, что обеспечивало бесперебойную поставку сырья.
В те годы в СССР насчитывалось около 40 гидролизных заводов, размещенных в различных регионах страны, включая Белоруссию, Казахстан, Молдавию, Грузию, Узбекистан. На территории Российской Федерации заводы действовали в центральных и северо-западных районах, в Поволжье, на Урале, в Сибири и на Дальнем Востоке. Они производили около 15 млн дал этилового спирта, более 1 млн т кормовых дрожжей и порядка 40 тыс. т фурфурола.
Кризисные 1990-е годы и развал отрасли. До начала 1990-х гг. гидролизное производство считалось крупной и высокорентабельной подотраслью микробиологической промышленности. При этом многие гидролизные предприятия являлись градообразующими и выполняли социально значимые функции (через собственные распределительные сети обеспечивали население городов и поселков теплом и водой, очищали коммунальные хозяйственно-бытовые стоки на производственных очистных сооружениях). Однако вскоре положение стало меняться. С распадом СССР прекратило существование Министерство микробиологической промышленности, формировавшее стратегию развития отрасли и представлявшее интересы гидролизных заводов на уровне союзного центра. На территории России осталось 17 заводов (из работавших в СССР 38), способных производить 20 млн дал этанола в год. В течение 1992 г. все они были преобразованы в акционерные общества различного типа. В федеральную собственность отошло 49-51% акций, оставшаяся часть была распределена среди работников заводов.
Известия ИГЭА. 2009. № 3 (65)
Таким образом, государство формально продолжало считаться основным собственником заводов, но по существу устранилось от разработки стратегии развития отрасли в новых экономических условиях. Предприятия оказались наедине со своими проблемами. Начались перебои с поставками сырья. Ухудшились условия жизни работников. Так, после либерализации цен железнодорожники и лесозаготовители стали получать зарплату в 4-6 раз более высокую, чем рабочие гидролизных заводов. Постоянно снижалось производство спирта, а выпуск кормовых дрожжей уменьшился по сравнению с 1990 г. почти в 15 раз, фурфурола — в 5-8 раз.
К началу 2000-х гг. в России осталось только восемь работающих заводов, способных выпускать порядка 5 млн дал этилового спир-
та. При этом на долю действующих заводов Иркутской области (в то время Тулунского и Бирюсинского) приходилась почти половина российского производства этанола. Вскоре началась череда банкротств, и в 2006 г. отечественная гидролизная промышленность прекратила свое существование.
Перспективы отрасли связаны с необходимостью наращивать производство биоэтанола, используемого в качестве моторного топлива. Во всем мире данное направление динамично развивается. Многие страны, включая Китай, приняли долгосрочные программы по увеличению производства биотоплива первого (из пищевого сырья) и второго (из древесных и сельскохозяйственных отходов) поколений. России также необходимо учитывать общемировые тенденции развития данной отрасли промышленности.
УДК 338(57) A.B. САМАРУХА
ББК 65.9(2Рос) зам. директора по региональной экономике
НИИ саморегулирования и развития предпринимательства Байкальского государственного университета экономики и права, кандидат экономических наук, доцент, г. Иркутск e-mail: [email protected]
НАПРАВЛЕНИЯ СТРАТЕГИЧЕСКОГО ПЛАНИРОВАНИЯ МЕЖМУНИЦИПАЛЬНОГО, МЕЖРЕГИОНАЛЬНОГО И МЕЖДУНАРОДНОГО СОТРУДНИЧЕСТВА И ВЗАИМОДЕЙСТВИЯ В БАЙКАЛЬСКОМ РЕГИОНЕ*
Определены основные тенденции стратегического планирования социально-экономического развития России и регионов. Отмечена и обоснована необходимость активного развития межмуниципальных, межрегиональных, а также международных отношений. Рассмотрены тенденции развития Байкальского региона. Предложены основные направления стратегического развития межмуниципальных, межрегиональных и международных отношений Иркутской области.
Ключевые слова: стратегия, социально-экономическое развитие, кризис, экология, межмуниципальное сотрудничество, международный аспект.
В современной ситуации нарастания негативном развитии геополитических меж-
экономических и социальных кризисных дународных отношений особенно необходи-
проявлений, при нестабильном и во многом ма разработка грамотной стратегии соци-
* Печатается при поддержке проекта РФФИ 08-06-98022 р_сибирь_а «Разработка стратегических направлений устойчивого развития Байкальского региона». Тема зарегистрирована во ВНТИЦентре, номер госрегис-трации 0120.0 808716.
© А.В. Самаруха, 2009
Сравнение гидролизной ферментации и биосинтеза газификации
[11] Харви Ф., Моррисон К., Маллиган М. Рост стоимости биотоплива снизился на
из-за резкого роста цен на зерно. Financial Times (Лондон), http: // www.
lexisnexis.com/us/lnacdemic/delivery/; 23 февраля 2007 г.
Лондонское издание.
[12] Кио М. Стабильная по мере роста. The Weekly Times
(Австралия), http://www.lexisnexis.com/us/lnacdemic/
delivery /; 6 января 2007 г.
[13] Lamb GM.Биотопливо многообещающе, но также создает проблемы.
Christian Science Monitor, http://www.lexisnexis.com/us/
lnacdemic / delivery /; 10 мая 2007 г.
[14] Маккарти М. Является ли этанол ответом на мировые нефтяные проблемы
или зеленой несбыточной мечтой. Лондон: Индепендент,
http://www.lexisnexis.com/us/lnacdemic/delivery/; 2008.
[15] Объединенные региональные информационные сети ООН. Африка; еда
поесть или запустить машину? Новости Африки, http: // www.lexisnexis.
ru / us / lnacdemic / delivery /; 23 октября 2007 г.
[16] Фон Браун Дж. Рай для горючего имеет свою цену. The Weekly
Times (Австралия), http://www.lexisnexis.com/us/lnacdemic/
delivery /; 22 августа 2007 г.
[17] Клэйтон М. В том, что глобальные цены на продукты питания растут, виновато ли биотопливо?
The Christian Science Monitor, http://www.csmonitor.com/
2008/0128 / p03s03-usec.htm; 2008.
[18] Дейл Р.Т., Тайнер В.Е. Экономико-технический анализ сухого помола этанола
: описание модели.Отчет № 06-04.
Вест-Лафайет, Индиана: Департамент экономики сельского хозяйства,
Университет Пердью; 2006.
[19] Reith JH, den Uil H, van Veen H, de Jong E, Elbersen HW,
Weusthuis R, et al. Совместное производство биоэтанола, электроэнергии,
и тепла из остатков биомассы. Отчет № ECN-RX-02-030.
Петтен, Нидерланды: Центр энергетических исследований
Нидерланды — Отделение биомассы; 2003.
[20] Wilkins MR, Widmer WW, Grohman K.Ферментативный гидролиз
кожуры грейпфрута для производства этанола и других продуктов.
Документ № 057047. Сент-Джозеф, Мичиган: ASABE; 2005.
[21] Вайман К., редактор. Справочник по биоэтанолу: производство и использование
. Вашингтон, округ Колумбия: Тейлор и Фрэнсис; 1996.
[22] Вэй Л., Пордезимо Л.О., Бэтчелор В.Д. Производство этанола
из древесины: сравнение гидролизной ферментации и биосинтеза газификации
. Бумага № 076036. St. Joseph, MI:
ASABE; 2007 г.
[23] Пиментел Д., Пацек Т.В. Производство этанола с использованием кукурузы,
проса, древесины; производство биодизеля с использованием сои и подсолнечника
. Nat Resour Res 2005; 14: 65–76.
[24] Го Дж, Сунь Й, Гребнер Д.Л. Использование лесной биомассы
для производства энергии в США: статус,
проблемы и государственная политика. Int Forestry Rev 2007; 9:
748–58.
[25] Иранмахбуб Дж., Надим Ф., Монеми С. Оптимизация кислотного гидролиза
: критический этап производства этанола из смешанной древесной щепы
.Биомасса Биоэнергетика 2002; 22: 401–4.
[26] Ли Й, Чен П., Ма Х. Влияние физических обработок на ферментативный гидролиз
кукурузной соломы. Документ № 016155. St.
Джозеф, Мичиган: ASAE; 2001.
[27] Ферраро Ф., Крафт М., Пенка К., Шарп Дж., Войлз Д., Кадам К. и др.
Технико-экономическое обоснование получения этанола из биомассы хвойных пород. Отчет №
NREL / SR-510-27310. Голден, Колорадо: Национальная лаборатория возобновляемых источников энергии
; 2004.
[28] Ли И, Руан Р., Чен П.Л., Лю З., Пан X, Линь X и др.Ферментативный
гидролиз кукурузной соломы, предварительно обработанной комбинированным разбавленным раствором
щелочью и гомогенизацией. Trans ASAE 2004;
47: 521–5.
[29] Чой Ч., Мэтьюз А.П. Кинетика двухступенчатого кислотного гидролиза в
осахаривании низкосортной биомассы: 1. Экспериментальные исследования
образования и разложения сахаров.
Биоресур Технол 1996; 58: 101–6.
[30] Шарма С.К., Калра К.Л., Гревал ХС. Ферментативное рубцевание
предварительно обработанных стеблей подсолнечника.Биомасса Биоэнергетика 2002; 23:
237–43.
[31] Содерстром Дж., Пилчер Л., Гальбе М., Закки Г. Двухступенчатая обработка паром
Предварительная обработка древесины мягких пород разбавленным H
2
SO
4
пропитка для производства этанола
. Биомасса Биоэнергетика 2003; 24: 475–86.
[32] Макалун А., Тейлор Ф., Йи В., Ибсен К., Вули Р. Определение
стоимости производства этанола из кукурузного крахмала и
лигноцеллюлозного сырья. Отчет №NREL / TP-580-28893.
Голден, Колорадо: Национальная лаборатория возобновляемых источников энергии; 2000.
[33] Duff SJB, Murrayh WD. Биоконверсия лесных продуктов
Промышленные отходы целлюлозы в этанол: обзор. Биоресур
Технол 1996; 55: 1–33.
[34] Wooley R, Ruth M, Sheehan J, Ibsen K, Majdeski H, Galvez A.
Разработка процесса получения этанола из лигноцеллюлозной биомассы и
экономичное использование прямоточного предварительного гидролиза разбавленной кислоты и
ферментативный гидролиз текущий и футуристический сценарии.
Отчет № NREL / TP-580-26157. Голден, Колорадо: Национальная лаборатория возобновляемых источников энергии
; 1999.
[35] Датар Р.П., Шенкман Р.М., Катени Б.Г., Хунке Р.Л., Льюис Р.С.
Ферментация производственного газа биомассы до этанола.
Biotechnol Bioeng 2004; 86: 587–94.
[36] Ху Дж., Ван И, Цао С., Эллиотт, округ Колумбия, Стивенс Д. Д., Уайт Дж. Ф..
Конверсия синтез-газа из биомассы в спирты и оксигенаты C2
с использованием Rh-катализаторов на носителе в микроканальном реакторе
.Катализ сегодня 2006; 120: 90–5.
[37] Классон К.Т., Акерсон М.Д., Клаузен Э.С., Гэдди Дж.Л.
Биоконверсия синтез-газа в жидкое или газообразное топливо.
Enzyme Microb Technol 1992; 14: 602–8.
[38] ван Кастерен Дж.М.Н., Диздаревич Д., Валл В.Р., Гуо Дж.,
Верберн Р. Биоэтанол из синтез-газа. Отчет № 0456372-R02.
Эйндховен, Нидерланды: Эйндховенский университет
Технологии, Телос и Ингения Консультанты и инженеры;
2005.
[39] Younesi H, Najafpour G, Mohamed AR. Этанол и ацетат
Производство синтез-газа в процессе ферментации
с использованием анаэробной бактерии Clostridium ljungdahlii. Biochem
Eng J 2005; 27: 110–9.
[40] Vega JL, Clausen EC, Gaddy JL. Проектирование биореакторов ферментации синтез-газа угля
. Рес Консерв Ресайклинг 1990; 3:
149–60.
[41] Французская школа химического машиностроения Т. Дэйва С. Суолма,
Государственный университет Миссисипи.Личное общение; 19
Март 2007 г.
[42] Процесс энергетической биоэнергетики BRI — краткое изложение технологии. BRI
Energy Inc., http://www.brienergy.com/; 2008.
[43] Вей Л., Томассон Дж., Брика Р.М., Бэтчелор В.Д.,
Колумбус Е.П., Вутен Дж. Экспериментальное исследование газогенератора с нисходящим потоком
. Документ № 066029. Сент-Джозеф, Мичиган: ASAE; 2006.
[44] Taconi KA. Метаногенное производство биогаза из
сточных вод системного газового брожения.Кандидат наук. диссертация.
Школа химической инженерии Дэйва С. Суолма, Миссисипи
Государственный университет; 2004.
[45] Рао М.С., Сингх С.П., Содха М.С., Дубей А.К., Шьям М.
Анализ стехиометрического, массового, энергетического и энергетического баланса
противоточной газификации с неподвижным слоем постпотребителей
остатков. Биомасса Биоэнергетика 2004; 27: 155–71.
[46] Eriksson G, Kjellstro
m B, Lundqvist B, Paulrud S. Сжигание
остатков гидролиза древесины в порошковой горелке мощностью 150 кВт.Топливо
2004; 83: 1635–41.
[47] Хенстра AM, Сипма Дж., Ринзема А., Стамс AJM. Микробиолой
ферментации синтез-газа для производства биотоплива. Curr Opin
Biotechnol 2007; 18: 200–6.
[48] Gaddy JL, Clausen EC. Clostridium ljungdahlii, анаэробный микроорганизм, продуцирующий этанол и ацетат
. Патент США
№ 612221; 1992.
[49] Ford JW. Производство уксусной кислоты путем ферментации синтез-газа
.РС. дипломная работа, Химическая школа Дэйва С. Суолма
Инженерное дело, Государственный университет Миссисипи; 2004.
[50] Коттер JL. Производство этанола и ацетата из синтез-газа
с использованием микробных катализаторов. Магистерская диссертация, Отдел
биомассы и биоэнергетики 33 (2009) 255–266 265
Процесс производства лигнина и летучих соединений из гидролизной жидкости | Биотехнология для производства биотоплива
Свойства древесной щепы
В таблице 1 приведен химический состав еловой щепы.Видно, что древесная щепа содержала 17,0, 47,2, 3,9, 2,8 и 26,5 мас.% Гемицеллюлозы, целлюлозы, экстрактивных веществ, растворимого в кислоте лигнина и нерастворимого в кислоте лигнина соответственно. Аналогичные составы древесной щепы ели описаны в других сообщениях [31, 32].
Таблица 1 Химический состав еловой щепы, приведенный в различных отчетахГидролизный анализ
В таблице 2 перечислены свойства гидролизной жидкости. Гидролизный раствор, полученный в этой работе, имел кислый pH 3.2–3.6, подразумевая, что ацетильные группы, прикрепленные к древесной щепе, были частично удалены и преобразованы в уксусную кислоту в процессе гидролиза [4, 33]. Очевидно, что при повышении температуры концентрации моносахаров и гидролизного лигнина увеличивались, в то время как концентрация полисахаров снижалась. Уменьшение содержания полисахаров было приписано разложению полисахаров, которое привело к образованию большего количества моносахаров в гидролизной жидкости (таблица 2) [25]. В другом сообщении увеличение температуры гидролиза тополя со 170 до 190 ° C при времени пребывания 60 мин немного увеличивало удаление моносахаров с 2.От 4 до 4,4% [33]. Результаты также показали, что молекулярная масса полисахаров была немного снижена за счет интенсификации жесткости процесса. Подобный феномен был отмечен другими исследователями, которые указали, что это уменьшение можно объяснить расщеплением полисахаридных цепей при повышенных температурах [25] или при длительном резидентстве [24]. Интересно, что содержание фурфурола значительно увеличивается с повышением температуры, что согласуется с результатами, опубликованными ранее [33]. В одном исследовании увеличение производства фурфурола из гидролизной жидкости процесса растворения целлюлозы на основе крафт-целлюлозы наблюдалось за счет повышения температуры со 170 до 190 ° C и увеличения времени пребывания с 20 до 100 минут [34].
Таблица 2 Свойства гидролизных и содовых растворов при различных температурах и времени пребыванияРезультаты в таблице 2 также показывают следующее: (1) при повышении температуры при времени гидролиза 15 мин концентрация уксусной кислоты увеличивалась; (2) при повышении температуры при времени гидролиза 45 мин концентрация уксусной кислоты уменьшалась; и (3) за счет увеличения времени гидролиза с 15 до 45 мин содержание уксусной кислоты в гидролизном растворе было уменьшено.Предполагается, что при коротком времени гидролиза, составляющем 15 мин, большее количество ацетильных групп отщеплялось при повышении температуры, и, таким образом, в гидролизной жидкости образовывалось больше уксусной кислоты [4]. При увеличении времени или в основном обеспечении более жестких условий гидролиза образовавшаяся уксусная кислота могла разложиться на другие продукты, и, следовательно, концентрация уксусной кислоты была снижена (таблица 2).
Молекулярная масса лигнина в гидролизной жидкости варьировалась от 21000 до 24000 г / моль.Было обнаружено, что молекулярная масса измельченного древесного лигнина, который был представителем природного лигнина для древесины ели [35], составляла около 23 500 г / моль [36].
Результаты также показали, что при увеличении времени с 15 до 45 минут наблюдалось небольшое увеличение концентраций фурфурола и моносахаров, но небольшое снижение концентраций полисахаров. Можно утверждать, что при увеличении времени полисахариды гидролизовались (т.е. расщеплялись) до моносахаров, но впоследствии моносахара были преобразованы в фурфурол [33].Концентрация лигнина незначительно изменялась с увеличением времени, что означает, что условия гидролиза не были сильными для удаления основного лигнина из древесной щепы [5].
Как обсуждалось ранее, некоторые соединения лигнина образовывались и осаждались / адсорбировались на поверхности варочного котла, которые собирались обработкой содой. Концентрация лигнина содового щелока в содовом растворе также указана в таблице 2. В содовом растворе не было обнаружено фурфурола, уксусной кислоты или сахаров, что указывает на то, что эти соединения не адсорбируются / не взаимодействуют с лигнином содового щелока и / или осаждаются в сосуд, но действительно остались в гидролизной жидкости.Очевидно, что при повышении температуры от 170 до 180 ° C из древесной щепы удалялось больше лигнина содового щелока, но при 190 ° C масса удаленного лигнина содового щелока была немного снижена. Результаты в таблице 2 показывают, что молекулярная масса лигнина содового щелока составляла около 3000 г / моль. Молекулярная масса лигнина натрового щелока была ниже, чем у гидролизного лигнина (таблица 2), а также у диоксанлигнина или лигнина, гидролизованного уксусной кислотой (6000 г / моль), о которых сообщалось в другом месте [37]. Молекулярная масса лигнина содового щелока была ниже при времени гидролиза 45 мин (по сравнению с 15 мин).Как правило, при увеличении времени из древесной щепы можно удалить больше лигнина. Снижение молекулярной массы может указывать на то, что удлинение по времени расщепляет часть межмолекулярных связей LCC и разрушает некоторые углеводы. Следовательно, общая молекулярная масса лигнина, присутствующего в содовом растворе, была снижена. Уменьшение количества лигнина в содовом растворе при 190 ° C может служить подтверждением этой гипотезы.
Подкисление гидролизных и содовых щелоков
В таблице 3 перечислены свойства гидролизных и содовых щелоков после подкисления (pH 1.5). Сравнение результатов в таблицах 2 и 3 показывает, что подкисление незначительно повлияло на концентрацию сахаров и гидролизный лигнин, подтверждая, что подкисление было неэффективным при извлечении лигнина или сахаров из гидролизной жидкости, и это явление было связано с высокой растворимостью гидролизного лигнина и сахара в кислых растворах. Выделение сахаров в осадках, полученных в результате подкисления гидролизного раствора, полученного при 180 ° C и 45 мин, было связано с более высокой концентрацией полисахаров в гидролизном растворе в этих условиях (таблица 2).В другой работе подкисление гидролизной жидкости в процессе производства растворяющейся целлюлозы на основе крафт-бумаги до pH 2 привело к незначительному разделению лигнина и сахара [15], но к снижению молекулярной массы полисахаров из-за гидролиза [38]. Сравнение концентраций фурфурола и уксусной кислоты до и после подкисления (таблицы 2, 3) показывает падение их концентраций после подкисления, что означает, что эти соединения были либо удалены из гидролизной жидкости [12], либо разложены в результате подкисления.Результаты в таблице 3 также показывают, что концентрация лигнина содового щелока значительно снизилась после доведения pH до 1,5.
Таблица 3 Свойства подкисленного гидролизного раствора и содового щелокаКак правило, по мере растворения большего количества лигнина и сахара в гидролизных растворах и образовании содовых растворов за 45 минут гидролизной обработки эти образцы были отобраны для дальнейшего анализа в этой работе. В таблице 4 показаны функциональные группы, присоединенные к осадкам, полученным в результате подкисления гидролизных и содовых растворов.
Таблица 4 Функциональная группа, связанная с осадками, образовавшимися в результате подкисления гидролиза, и содовыми растворами, образовавшимися после 45 мин обработки гидролизомАлифатические гидроксильные (ОН), фенолятные и карбоксилатные группы изначально существуют в лигнине [30, 39], а некоторые свободные фенолятные и алифатические группы могут образовываться в процессе гидролиза [40, 41]. Образование связей C – C должно происходить в реакции конденсации в кислых условиях и вносить вклад в связи C – C (Таблицы 2, 3) [42, 43].Хотя таблица 4 показывает уменьшение замещения C-5, другие углерод-углеродные связи, такие как β-β и β-5, могут образовываться в реакции конденсации [44, 45]. Сообщалось, что осадки гидролизного раствора, полученные в результате парового гидролиза смешанной древесины лиственных пород, содержали 1,88 ммоль / г алифатических гидроксильных групп, что согласуется с данными настоящей работы [41].
Из таблицы 4 также видно, что повышение температуры гидролиза снижает количество функциональных групп в осадках.Уменьшение содержания гваяцильных, фенолятных и карбоксилатных групп в осадках (таблица 4) согласуется с уменьшением молекулярной массы при высокой температуре, показанным в таблице 2. В предыдущем исследовании утверждалось, что при повышении температуры разложение карбоксилатных групп в древесине ели увеличилось [46], что согласуется с деградацией карбоксилатных групп в данной работе при повышении температуры от 170 до 190 ° C. Результаты в таблице 4 также показывают, что осадки содового раствора обычно имеют меньше алифатических и гваяцилгидроксильных групп, но больше замещенных C5, карбоксилатных и p -гидроксифенильных групп.
Термические свойства осадков из гидролизной жидкости
Термический анализ осадков был проведен для определения их конечного применения. Потеря массы осадков, образовавшихся при подкислении гидролизной жидкости, показана как функция температуры на фиг. 2а. Видно, что потеря массы осадков была значительной, и только 20 мас.% Осадков осталось в виде полукокса при 700 ° C. Скорость потери массы образцов показала сходное поведение разложения для всех образцов в диапазоне температур 100–700 ° C (рис.2б). В литературе сообщается, что пиролиз лигнина приводит к получению 60 мас.% Полукокса [47, 48]. Известно, что поведение материалов при пиролизе зависит от их состава и структуры [49]. Непрерывное резкое разложение осадков можно объяснить двумя причинами. (1) Осадки содержали летучие компоненты, которые постепенно испарялись при повышенной температуре. Как указано выше, концентрация фурфурола в гидролизном растворе падает из-за подкисления, и это может косвенно указывать на то, что фурфурол осаждается в результате подкисления (таблицы 2, 3).Испарение фурфурола из осадков будет рассматриваться как эндотермический эффект при термическом анализе и способствовать тенденции потери веса, наблюдаемой на рис. 2. (2) Лигнин разлагался в присутствии неорганических соединений при более высокой температуре. Сообщалось, что серная кислота усиливает разложение лигнина при пиролизе [50]. Поскольку осадки образовывались в результате обработки гидролизной жидкости серной кислотой, возможно, что некоторые молекулы серной кислоты адсорбировались на осадках и, следовательно, способствовали тенденции, наблюдаемой на рис.2.
Рис. 2Анализ потери массы ( a ) и скорость потери массы ( b ) осадков, образовавшихся в результате подкисления гидролизных жидкостей, образовавшихся при 170, 180 и 190 ° C
Чтобы лучше понять характеристики осадков, их термические свойства были проанализированы с помощью ДСК. Тепловой поток осадков, образующихся при подкислении гидролизной жидкости при 170 ° C, показан на рис. 3. Пики кривой нагрева ниже 200 ° C, температура кипения уксусной кислоты составляет 118 ° C, а фурфурола — 161.7 ° С [51]. Таким образом, резкое падение кривых ДСК выше 150 ° C можно объяснить испарением фурфурола из осадков. Аналогичный результат был получен при анализе DSC осадков, образовавшихся в результате подкисления гидролизной жидкости, образовавшейся при 180 или 190 ° C (не показано). Из рис. 2 и 3 видно, что осадки, полученные в результате подкисления гидролизной жидкости, содержат летучие соединения, которые ухудшают термические свойства осадков таким образом, что они не подходят в качестве биотоплива.
Рис. 3Анализ DSC осадков, образовавшихся при подкислении образца 1; гидролиз при 170 ° C, 15 мин; синяя кривая показывает нагрев по сравнению с красной кривой , которая показывает охлаждение
Термические свойства лигнина неочищенного содового щелока
На рисунке 4 показаны потери веса и скорость потери веса неочищенного лигнина содового щелока, полученного в результате подкисления содового щелока. Видно, что первый пик возник в одном и том же положении для всех трех температур, в то время как второй пик разложения произошел при более высокой температуре для образца, приготовленного при 190 ° C гидролиза, что указывает на то, что осадки были бы более термически стабильными, если бы они были получены при более высокой температуре в процессе гидролиза.Таблица 4 показывает, что общие количества функциональных групп составляют 3,43, 3,17 и 2,18 ммоль / г для осадков, полученных в результате гидролиза при 170, 180 и 190 ° C, соответственно. В более раннем исследовании пиролиз древесины при температуре выше 200 ° C приводил к деградации карбоксилатных групп [46]. Точно так же настоящая работа показывает, что высокая концентрация функциональных групп приводит к большой потере массы при пиролизе.
Рис. 4Потеря массы ( a ) и скорость потери массы ( b ) неочищенного лигнина содового щелока, собранного при различных температурах гидролиза (170, 180 и 190 ° C), но при 45 мин.
На рис. 5 показан тепловой поток осадков, образовавшихся при подкислении содового щелока.Все осадки показали пик минимального теплового потока (то есть точку плавления) примерно при 220–230 ° C. Во время подкисления содового щелока серная кислота реагирует с присутствующим NaOH и образует сульфит натрия (Na 2 SO 3 ) и сульфат натрия (Na 2 SO 4 ). Эти соли также будут выпадать в осадок вместе с лигнином, присутствующим в содовом растворе.
Рис. 5Анализ методом ДСК осадков, образовавшихся в результате подкисления содового щелока, образовавшегося при различных температурах гидролиза и 45 мин.
Чтобы проверить, есть ли эндотермические пики на рис.5 (т.е. температура плавления) обусловлены плавлением неорганических солей в осадках, тепловые потоки чистых солей были проанализированы как функции температуры (дополнительный файл 1). Среди NaOH, Na 2 SO 3 и Na 2 SO 4 только Na 2 SO 4 показал пик на кривой нагрева приблизительно при 240–250 ° C. Температуры плавления NaOH, Na 2 SO 3 и Na 2 SO 4 составили 318,33.4 и 884 ° C соответственно [50]. Более раннее исследование показало, что эндотермический пик при температуре около 250 ° C был обусловлен изменениями кристаллической структуры Na 2 SO 4 [52]. Это подтвердило, что пик для Na 2 SO 4 был следствием изменений в структуре Na 2 SO 4 , присутствующего в лигнине содового щелока. Таким образом, результаты подтвердили, что пики теплового потока на рис.5 были созданы не органическими веществами осадков, а неорганическими соединениями (т.е.е. сульфат натрия), присутствующий в осадках.
Термические свойства очищенного лигнина содового щелока
Для удаления неорганических солей из лигнина содового щелока осадки, образовавшиеся при подкислении содового щелока, диализовали в воде с мембраной. На рис. 6а показана потеря веса осадков после диализа. Видно, что пиролиз осадков привел к получению 60 мас.% Полукокса, а осадки, полученные в результате гидролиза при 190 ° C, образовали меньше полукокса, чем осадки, полученные при более низкой температуре.На рис. 6b видно, что образцы, полученные гидролизом при 170 и 180 ° C, начали разлагаться при более низкой температуре 200 ° C; однако образец, полученный гидролизом при 190 ° C, разложился, когда температура была выше 300 ° C. Сравнение результатов на рис. 4 и 6 также показано, что после пиролиза конечное содержание полукокса было выше в очищенных осадках, чем в неочищенных осадках. Это говорит о том, что мембранный диализ удалил некоторые низкомолекулярные органические соединения в дополнение к неорганическим.Следовательно, после пиролиза беззольного лигнина образовалось больше полукокса.
Рис.6Потеря массы ( a ) и скорость потери массы ( b ) очищенного содового щелока, лигнина содового щелока, собранного при различных температурах гидролиза (170, 180 и 190 ° C), но через 45 мин.
Следует отметить, что потеря массы на рис. 6 (20–40 мас.%) Меньше типичной потери массы при пиролизе лигнина (40–50%). Как обсуждалось ранее, для удаления неорганических солей в процессе диализа некоторые соединения с малой молекулярной массой могли пройти через мембрану.Соответственно, очищенные осадки (т.е. очищенный лигнин натрового щелока) имели более высокую молекулярную массу и, таким образом, демонстрировали более высокую термическую стабильность (т.е. меньшую потерю массы).
Тепловой поток очищенных осадков также был проанализирован как функция температуры (дополнительный файл 1). Пик теплового потока наблюдался при 180–190 ° C, что свидетельствовало о том, что структура выделений изменила свое состояние из стеклообразного состояния в каучукоподобное. Tg лигнина зависит от процессов производства лигнина и от источника древесины [53].Сообщалось, что Tg лигнина, полученного в результате кислотного гидролиза древесины хвойных пород, составляла 95 ° C, в то время как Tg лигнина, полученного в результате парового взрыва древесины хвойных пород, составляла 139 ° C [54]. В другом сообщении Tg лигнина, полученного при гидролизе древесины лиственных пород горячей водой, составляла 170–180 ° C [55]. В этой работе увеличение температуры гидролизной обработки со 170 до 190 ° C привело к увеличению Tg с 184 до 192 ° C. Это согласуется с более ранним исследованием, в котором сообщалось об увеличении Tg на 8 ° C при повышении температуры обработки горячей водой древесины твердых пород на 20 ° C [55].Кроме того, анализ теплового потока очищенных осадков не выявил точки плавления между 200 и 220 ° C, что подтверждает, что точки плавления на фиг. 4 действительно были обусловлены присутствием неорганических солей в нечистых осадках.
Разработка процессов и применение продуктов
Следуя шагам, показанным на рис. 1, был разработан баланс масс, чтобы определить, насколько гидролиз горячей водой удалит различные компоненты из древесной щепы, и результаты приведены в таблице 5.Наблюдается, что примерно 25% древесной щепы было выделено путем гидролиза. В литературе указано, что при температуре 180 ° C и времени 2 часа растворяется больше компонентов, чем при более высокой температуре и более продолжительном времени в процессе гидролиза, что подтверждает результаты, представленные в таблице 5 [56]. Сумма органических соединений в гидролизном растворе после подкисления, осадках подкисления и лигнина содового щелока составляла примерно 95–98%, что свидетельствует о высокой достоверности анализа.Результаты также показывают, что подкисление гидролизного раствора снижает содержание уксусной кислоты и фурфурола в гидролизном растворе. Саид и др. заявили, что уксусная кислота и фурфурол составляют 1,31–1,52 и 0,18–0,31% от общего содержания твердых веществ в промышленно производимом гидролизном растворе древесины лиственных пород [4]. В настоящей работе содержание уксусной кислоты и фурфурола в гидролизном растворе мягкой древесины составляло от 0,5 до 0,7 мас.% И от 0,2 до 2,4 мас.% От общего содержания твердых веществ в гидролизном растворе, соответственно.Расхождение можно объяснить различиями в сырье, используемом в этих двух исследованиях. Следовательно, подкисление гидролизной жидкости (а не энергоемкое испарение) может быть предложено в качестве метода выделения летучих соединений. Однако большая часть полисахаров, моносахаров и гидролизного лигнина оставалась в растворе. Интересно отметить, что количество лигнина, адсорбированного на варочном котле (и, таким образом, удаленного с помощью содовой обработки), было выше, чем количество лигнина, удаленного с помощью подкисляющего гидролизного раствора.
Таблица 5 Масса компонентов в обработанной древесной щепе, осадках подкисления гидролизного раствора, подкисленном гидролизном растворе, подкисленном содовом щелоке и лигнине содового щелока (на 100 г высушенной в печи древесины)На рисунке 7 представлен процесс извлечения органических материалов из гидролизной жидкости.Результаты этой работы показывают, что гидролиз древесной щепы приводит к извлечению моно- и полисахаров, фурфурола, уксусной кислоты, а также лигнина из древесной щепы. Как указывалось ранее, некоторые менее растворимые соединения лигнина образовывались и адсорбировались на внутренней поверхности варочного котла. В крупномасштабной операции часть этих соединений будет адсорбирована на варочном котле. После насыщения поверхности варочного котла эти соединения не смогут адсорбироваться на варочном котле из-за напряжения сдвига и давления, приложенного в процессе гидролиза от подачи / разгрузки варочного котла, а также циркуляции гидролизной жидкости в процессе гидролиза, как это было проведено. в промышленности [5].Следовательно, эти соединения будут коагулироваться и образовывать хлопья соединения лигнина. Процесс осаждения / фильтрации может облегчить осаждение этих хлопьев лигнина из системы. Чтобы получить очищенные хлопья лигнина, неочищенный гидролизный лигнин, отделенный посредством седиментации / фильтрации, можно дополнительно промыть кислотами, например серной кислотой, а затем очистить с помощью мембраны. Органические вещества можно выделить из гидролизной жидкости путем нагревания и подкисления до pH 1,5. Утверждается, что нагревание является ключевым элементом в образовании и коагуляции крафт-лигнина в технологии LignoForce [16].Результаты в таблицах 2, 3 и 5 показывают, что подкисление гидролизного раствора можно использовать как средство для извлечения летучих соединений (например, фурфурола и уксусной кислоты) из гидролизного раствора с 2% осадков в расчете на массу древесины (таблица 5). .
Рис. 7Способ получения гидролизного лигнина и осадков
После этого подкисленный гидролизный раствор можно направлять на очистку сточных вод. Затем его можно нейтрализовать и дополнительно обработать с помощью процессов биологической очистки [57].В разработанном процессе, показанном на фиг. 7, отработанная кислота, которая образуется в процессе очистки лигнина, может использоваться в качестве источника кислоты для подкисления гидролизной жидкости. Этот процесс аналогичен LignoForce в том, что он использует резервуар для отстаивания / фильтрации для выделения соединений лигнина из гидролизной жидкости и использует кислотную промывку для дальнейшей очистки соединений лигнина.
Термические анализы подтверждают, что лигнин натрового щелока имел термостабильные характеристики. Следовательно, этот продукт может быть использован в производстве композитов (например,g .. композиты лигнин – эпоксидная смола), в которых термическая стабильность композита является преимуществом [58]. Однако для подтверждения этого требуется более глубокий анализ. Кроме того, подкисление гидролизной жидкости привело к образованию осадков, которые в основном представляли собой фурфурол и уксусную кислоту. Эти химикаты могут использоваться в качестве платформенных химикатов для производства других химикатов с добавленной стоимостью, таких как пластмассы, фармацевтическая и агрохимическая промышленность [59].
Как видно из Таблицы 5, можно было бы производить 1, 5 и 19 тонн фурфурола из 1000 тонн древесной щепы, если бы были соблюдены процедуры производства образцов 4, 5 и 6, соответственно.Уксусная кислота будет производиться в количестве 6, 4 и 3 тонны, если следовать процедурам для образцов 4, 5 и 6 соответственно. Следует отметить, что этот процесс может быть неэкономичным, если его рассматривать независимо, поскольку выход экстракции довольно низкий (Таблица 5). Однако гидролиз коммерчески используется в качестве стадии предварительной обработки при производстве растворяющейся целлюлозы после крафт-процесса и используется для производства древесных гранул [4–6]. Предлагаемый процесс может быть экономичным в этих процессах, так как он может генерировать дополнительные доходы от отходов.Однако необходимо провести подробный экономический анализ, чтобы понять финансовую осуществимость предлагаемого процесса.
Производство ароматных спиртов из древесины и возможные области применения для варки и спиртных напитков из древесины
В этой работе исследуется использование древесины для дорогостоящего производства новых алкогольных напитков и ликеров с натуральными ароматизаторами. В этом процессе используется наша оригинальная технология мокрого бисерного фрезерования (WBM), которая обеспечивает прямое ферментативное осахаривание и спиртовую ферментацию древесины без химической и термической обработки, что приводит к отсутствию токсичных соединений.Когда продукты на спиртовой основе из различных пород древесины, включая Cryptomeria japonica (кедр), Cerasus × yedoensis (вишня) и Betula platyphylla (береза), были проанализированы с помощью SPME-GC-MS, различные природные ароматические компоненты были обнаружены в каждом. Корреляционный анализ с использованием Heracles NEO и ASTREE V5 показал, что спирты из древесины имеют другие вкусовые и вкусовые характеристики по сравнению с существующими коммерческими спиртными напитками. В пилотных экспериментах был определен выход алкогольного напитка на количество биомассы.Пилотные опыты показали важность оптимального размера древесных частиц во время WBM для эффективного производства спирта. Хотя спирт, произведенный из дерева, должен быть сначала признан безопасным для употребления в пищу человеком, это первое описание питьевых спиртов, произведенных из дерева. Эта работа может открыть важные возможности для использования лесных ресурсов в производстве продуктов питания, чтобы улучшить нынешнее состояние лесного хозяйства.
Эта статья в открытом доступе
Подождите, пока мы загрузим ваш контент… Что-то пошло не так. Попробуйте снова?предварительная обработка, ферментативный гидролиз, ферментация, реология и смешивание
Необходимо разработать альтернативные источники энергии, чтобы исправить истощение запасов ископаемого топлива.Лигноцеллюлозная биомасса — это доступное во всем мире возобновляемое сырье, содержащее богатую сахарную платформу, которая может быть преобразована в биоэтанол путем соответствующей обработки. Ключевые этапы процесса, предварительная обработка, ферментативный гидролиз и ферментация, подверглись значительному количеству исследований и разработок за последние десятилетия, приближая процесс к коммерциализации. Чтобы коммерциализация была успешной, процесс должен работать при высоком содержании сухого вещества в биомассе, особенно на стадии ферментативного гидролиза, которая влияет на концентрацию этанола в конечном ферментационном бульоне.Биомасса превращается в густую пасту со сложной реологией для эффективного перемешивания. По мере увеличения консистенции биомассы увеличивается предел текучести, что ограничивает эффективность смешивания в обычных резервуарах с мешалкой. Цель этого обзора — представить особенности и перспективы переработки биомассы в этанол. Особое внимание уделяется реологии и смешиванию биомассы на стадии ферментативного гидролиза как одной из важнейших проблем в данной области.
Ссылки
[1] Виикари Л., Вехмаанперя Дж., Койвула А. Биомасса Биоэнергетика 2012, 46, 13–24. Искать в Google Scholar
[2] Hahn-Hägerdal B, Galbe M, Gorwa-Grauslund MF, Lidén G, Zacchi G. Trends Biotechnol. 2006, 24, 549–556. Искать в Google Scholar
[3] Schell DJ, Riley CJ, Dowe N, Farmer J, Ibsen KN, Ruth MF, Toon ST, Lumpkin RE. Биоресурсы. Technol. 2004, 91, 179–188. Искать в Google Scholar
[4] Larsen J, Petersen MØ, Thirup L, Li HW, Iversen FK. Chem. Англ. Technol. 2008, 31, 765–772. Искать в Google Scholar
[5] Tolan JS. Clean Technol. Environ. Политика 2002, 3, 339–345. Искать в Google Scholar
[6] Волынец Б., Дахман Ю. Межд. J. Energy Environ. 2011, 2, 427–446. Искать в Google Scholar
[7] Sticklen M. Curr. Opin. Biotechnol. 2006, 17, 315–319. Искать в Google Scholar
[8] Kim S, Dale BE. Биомасса Биоэнергетика 2004, 26, 361–375. Искать в Google Scholar
[9] Faraco V, Hadar Y. Обновить. Устойчивая энергетика Ред. 2011 г., 15, 252–266. Искать в Google Scholar
[10] Petersen MØ, Larsen J, Thomsen MH. Биомасса Биоэнергетика 2009, 33, 834–840. Искать в Google Scholar
[11] Zhang J, Chu D, Huang J, Yu Z, Dai G, Bao J. Biotechnol. Bioeng. 2010, 105, 718–728. Искать в Google Scholar
[12] Li Q, Gao Y, Wang H, Li B, Liu C, Yu G, Mu X. Bioresour. Technol. 2012, 125, 193–199. Искать в Google Scholar
[13] Saha BC, Yoshida T, Cotta MA, Sonomoto K. Ind. Crop. Prod. 2013, 44, 367–372. Искать в Google Scholar
[14] Ян Л., Цао Дж., Джин И, Чанг Х, Джамиль Х, Филлипс Р., Ли З. Биоресурсы. Technol. 2012, 124, 283–291. Искать в Google Scholar
[15] Amiri H, Karimi K, Zilouei H. Bioresour. Technol. 2014, 152, 450–456. Искать в Google Scholar
[16] Saha BC, Cotta MA. New Biotechnol. 2010, 27, 10–16. Искать в Google Scholar
[17] Хан М., Кан К. Э., Ким И, Чой Г. В.. Process Biochem. 2013, 48, 488–495. Искать в Google Scholar
[18] Benjamin Y, Cheng H, Görgens JF. Ind. Crop. Prod. 2013, 51, 7–18. Искать в Google Scholar
[19] Sundar S, Bergey NS, Salamanca-Cardona L, Stipanovic A, Driscoll M . Углеводы. Polym. 2014, 100, 195–201. Искать в Google Scholar
[20] Kumar S, Kothari U, Kong L, Lee YY, Gupta RB. Биомасса Биоэнергетика 2011, 35, 956–968. Искать в Google Scholar
[21] Ван З.Дж., Чжу Дж.Й., Залесный-младший Р.С., Чен К.Ф. Топливо 2012, 95, 606–614. Искать в Google Scholar
[22] Сантос Р. Б., Сокровище Т, Гонсалес Р., Филлипс Р., Ли Дж. М., Джамиль Х., Чанг Х. Биоресурсы. Technol. 2012, 117, 193–200. Искать в Google Scholar
[23] Sun Y, Cheng J. Bioresour. Technol. 2002, 83, 1–11. Искать в Google Scholar
[24] Балат М. Energy Convers. Управлять. 2011, 52, 858–875. Искать в Google Scholar
[25] Gírio FM, Fonseca C, Carvalheiro F, Duarte LC, Marques S, Bogel-ukasik R. Биоресурсы. Technol. 2010, 101, 4775–4800. Искать в Google Scholar
[26] Буранов А.Ю., Мазза Г. Ind. Crop. Prod. 2008, 28, 237–259. Искать в Google Scholar
[27] Vermerris W. In Генетическое улучшение биоэнергетических культур , 1-е изд. Вермеррис В., редактор. Springer Science and Business Media: Нью-Йорк, 2008, Vol. 1, стр. 89. Поиск в Google Scholar
[28] Sathitsuksanoh N, Xu B, Zhao B, Zhang Y-HP. PLoS One 2013, 8, 1–6.Искать в Google Scholar
[29] Fu C, Mielenz JR, Xiao X, Ge Y, Hamilton CY, Rodriguez M Jr, Chen F, Foston M, Ragauskas A, Bouton J, Dixon RA, Wang Z-Y. Proc. Natl. Акад. Sci. 2011, 108, 3803–3808. Искать в Google Scholar
[30] Shen H, Poovaiah CR, Ziebell A, Tschaplinski TJ, Pattathil S, Gjersing E, Engle NL, Katahira R, Pu Y, Sykes R, Chen F, Ragauskas AJ, Mielenz MG, Hahn , Дэвис М., Стюарт С. Н. младший, Диксон Р. А.. Biotechnol. Биотопливо 2013, 6, 71–86.Искать в Google Scholar
[31] Himmel ME, Ding S-Y, Johnson DK, Adney WS, Nimlos MR, Brady JW, Foust TD. Science 2007, 315, 804–807. Искать в Google Scholar
[32] Varga E, Réczey K, Zacchi G. Appl. Biochem. Biotechnol. 2004, 113–116, 509–523. Искать в Google Scholar
[33] Хейтц М., Чапек-Менар Э, Кёберле П.Г., Ганье Дж., Чорнет Э., Оверенд Р.П., Тейлор Дж. Д., Ю. Э. Биоресур. Technol. 1991, 35, 23–32. Искать в Google Scholar
[34] Ву М.М., Чанг К., Грегг Д.Д., Буссаид А., Битсон Р.П., Саддлер Дж. Заявл. Biochem. Biotechnol. 1999, 77–79, 47–54. Искать в Google Scholar
[35] Avci A, Saha BC, Dien BS, Kennedy GJ, Cotta MA. Биоресурсы. Technol. 2013, 130, 603–612. Искать в Google Scholar
[36] Zhu Y, Lee YY, Elander RT. Заявл. Biochem. Biotechnol. 2005, 121–124, 1045–1054. Искать в Google Scholar
[37] Kootstra AMJ, Beeftink HH, Scott EL, Sanders JPM. Biotechnol. Биотопливо 2009, 2, 31–45. Искать в Google Scholar
[38] Mosier N, Hendrickson R, Ho N, Sedlak M, Ladisch MR. Биоресурсы. Technol. 2005, 96, 1986–1993. Искать в Google Scholar
[39] Wang Z, Cheng JJ. Energy Fuels 2011, 25, 1830–1836. Искать в Google Scholar
[40] Kim S, Holtzapple MT. Биоресурсы. Technol. 2005, 96, 1994–2006. Искать в Google Scholar
[41] Hu Z, Wen Z. Biochem. Англ. J. 2008, 38, 369–378. Искать в Google Scholar
[42] МакИнтош С., Ванков Т. Биомасса Биоэнергетика 2011, 35, 3094–3103.Искать в Google Scholar
[43] Кан К. Э., Чон Г-Т, Суну К., Пак Д-Х. Биопроцесс Биосист. Англ. 2012, 35, 77–84. Искать в Google Scholar
[44] Теймури Ф., Лауреано-Перес Л., Ализаде Х., Дейл Б.Э. Биоресурсы. Technol. 2005, 96, 2014–2018 гг. Искать в Google Scholar
[45] Варга Э., Шмидт А.С., Рече К., Томсен А.Б. Заявл. Biochem. Biotechnol. 2003, 104, 37–50. Искать в Google Scholar
[46] García-Cubero MT, González-Benito G, Indacoechea I, Coca M, Bolado S .Биоресурсы. Technol. 2009, 100, 1608–1613. Искать в Google Scholar
[47] Banerjee G, Car S, Scott-Craig JS, Hodge DB, Walton JD. Biotechnol. Биотопливо 2011, 4, 16–31. Искать в Google Scholar
[48] Ли К., Книрим Б., Маниссери С., Арора Р., Шеллер Х. В., Ауэр М., Фогель К. П., Симмонс Б. А., Сингх С. Биоресур. Technol. 2010, 101, 4900–4906. Искать в Google Scholar
[49] Sathitsuksanoh N, Zhu Z, Zhang Y-HP. Целлюлоза 2012, 19, 1161–1172.Искать в Google Scholar
[50] Park N, Kim H-Y, Koo B-W, Yeo H, Choi I-G. Биоресурсы. Technol. 2010, 101, 7046–7053. Искать в Google Scholar
[51] Kim KH, Hong J. Bioresour. Technol. 2001, 77, 139–144. Искать в Google Scholar
[52] Bitra VSP, Womac AR, Igathinathane C, Miu PI, Yang YT, Smith DR, Chevanan N, Sokhansanj S. Bioresour. Technol. 2009, 100, 6578–6585. Искать в Google Scholar
[53] Bitra VSP, Womac AR, Chevanan N, Miu PI, Igathinathane C, Sokhansanj S, Smith DR .Пудра Технол. 2009, 193, 32–45. Искать в Google Scholar
[54] Sant’Ana da Silva A, Inoue H, Endo T, Yano S, Bon EPS. Биоресурсы. Technol. 2010, 101, 7402–7409. Искать в Google Scholar
[55] Yu Z, Zhang B, Yu F, Xu G, Song A. Bioresour. Technol. 2012, 121, 335–341. Искать в Google Scholar
[56] Rabelo SC, Filhho RM, Costa AC. Заявл. Biochem. Biotechnol. 2009, 153, 139–150. Искать в Google Scholar
[57] Schmidt AS, Thomsen AB. Биоресурсы. Technol. 1998, 64, 139–151. Искать в Google Scholar
[58] Сильверштейн Р.А., Чен Й., Шарма-Шиваппа Р.Р., Бойетт, М.Д., Осборн Дж. Биоресур. Technol. 2007, 98, 3000–3011. Искать в Google Scholar
[59] Ямасита Ю., Шоно М., Сасаки К., Накамура Ю. Углеводы. Polym. 2010, 79, 914–920. Искать в Google Scholar
[60] Diaz A, Toullec JL, Blandino A, Ory I., Caro I. Chem. Англ. Пер. 2013, 32, 949–954. Искать в Google Scholar
[61] Li Q, He Y-C, Xian M, Jun G, Xu X, Yang J-M, Li L-Z. Биоресурсы. Technol. 2009, 100, 3570–3575. Искать в Google Scholar
[62] Li B, Asikkala J, Filpponen I, Argyropoulos DS. Ind. Eng. Chem. Res. 2010, 49, 2477–2484. Искать в Google Scholar
[63] Zhang Y-HP, Ding SY, Mielenz JR, Cui JB, Elander RT, Laser M, Himmel ME, McMillan JR, Lynd LR. Biotechnol. Bioeng. 2007, 97, 214–223. Искать в Google Scholar
[64] Zhao X, Cheng K, Liu D. Appl. Microbiol. Biotechnol. 2009, 82, 815–827.Искать в Google Scholar
[65] Sannigrahi P, Miller SJ, Ragauskas AJ. Углеводы. Res. 2010, 345, 965–970. Искать в Google Scholar
[66] Арак Э., Парра С., Фрир Дж., Контрерас Д., Родригес Дж., Мендонка Р., Баеза Дж. Enzyme Microb. Tech. 2008, 43, 214–219. Искать в Google Scholar
[67] Гу Т. В Предварительная обработка зеленой биомассы для производства биотоплива , Гу Т., редактор. Springer: Нидерланды, 2013, т. 1, p 107. Искать в Google Scholar
[68] Narayanaswamy N, Faik A, Goetz DJ, Gu T. Биоресурсы. Technol. 2011, 102, 6995–7000. Искать в Google Scholar
[69] Бак Дж. С., Ко Дж. К., Чой Ай-Джи, Парк Й-К, Сео Дж-Х, Ким К. Х. Biotechnol. Bioeng. 2009, 104, 471–482. Искать в Google Scholar
[70] Malherbe S, Cloete TE. Rev. Environ. Sci. Bio. 2002, 1, 105–114. Искать в Google Scholar
[71] Keller FA, Hamilton JE, Nguyen QA. Заявл. Biochem. Biotechnol. 2003, 105–108, 27–41. Искать в Google Scholar
[72] Tao L, Aden A, Elander RT, Pallapolu VR, Lee YY, Garlock RJ, Balan V, Dale BE, Kim Y, Mosier NS, Ladisch MR, Falls M, Holtzapple MT, Sierra R , Ши Дж., Эбрик М.А., Редмонд Т., Ян Б., Вайман К.Э., Хамес Б., Томас С., Уорнер РЭ. Биоресурсы. Technol. 2011, 102, 11105–11114. Искать в Google Scholar
[73] Кази Ф.К., Фортман Дж. А., Анекс Р.П., Хсу Д.Д., Аден А., Дутта А., Котандараман Г. Fuel 2010, 89, S20 – S28. Искать в Google Scholar
[74] Eggeman T, Elander RT. Биоресурсы. Technol. 2005, 96, 2019–2025. Искать в Google Scholar
[75] Teeri TT. Trends Biotechnol. 1997, 15, 160–167. Искать в Google Scholar
[76] Dashtan M, Schraft H, Qin W. Внутр. J. Biol. Sci. 2009, 5, 578–595. Искать в Google Scholar
[77] Kuhad RC, Gupta R, Singh A. Enzyme Res. 2011, 2011, 1–10. Искать в Google Scholar
[78] Shallom D, Shoham Y. Curr. Opin. Microbiol. 2003, 6, 219–228. Искать в Google Scholar
[79] Väljamäe P, Kipper K, Pettersson G, Johansson G. Biotechnol. Bioeng. 2003, 84, 254–257. Искать в Google Scholar
[80] Zhang M, Su R, Qi W, He Z. Appl.Biochem. Biotechnol. 2010, 160, 1407–1414. Искать в Google Scholar
[81] Селиг MJ, Knoshaug EP, Adney WS, Himmel ME, Decker SR. Биоресурсы. Technol. 2008, 99, 4997–5005. Искать в Google Scholar
[82] Йе Л, Су Х, Шмитц Г.Е., Мун ЙХ, Чжан Дж., Маки Р.И., Канн IKO. Заявл. Eviron. Microbiol. 2012, 78, 7048–7059. Искать в Google Scholar
[83] Zhu L, O’Dwyer JP, Chang VS, Granda CB, Holtzapple MT. Биоресурсы. Technol. 2008, 99, 3817–3828.Искать в Google Scholar
[84] Chang VS, Holtzapple MT. Заявл. Biochem. Biotechnol. 2000, 84–86, 5–37. Искать в Google Scholar
[85] O’Dwyer JP, Zhu L, Granda CB, Holtzapple MT. Биоресурсы. Technol. 2007, 98, 2969–2977. Искать в Google Scholar
[86] Zhang YH, Lynd LR. Biotechnol. Bioeng. 2004, 88, 797–824. Искать в Google Scholar
[87] Ye Z, Hatfield KM, Berson RE. Биоресурсы. Technol. 2012, 106, 133–137.Искать в Google Scholar
[88] Kumar R, Wyman CE. Biotechnol. Bioeng. 2009, 102, 1544–1557. Искать в Google Scholar
[89] Eriksson T, Börjesson J, Tjerneld F. Enzyme Microb. Tech. 2002, 31, 353–364. Искать в Google Scholar
[90] Стил Б., Радж С., Нгием Дж., Стоуэрс М. Прил. Biochem. Biotechnol. 2005, 121–124, 901–910. Искать в Google Scholar
[91] Weiss N, Börjesson J, Pedersen LS, Meyer AS. Biotechnol.Биотопливо 2013, 6, 5–19. Искать в Google Scholar
[92] Lee D, Yu AHC, Saddlert JN. Biotechnol. Bioeng. 1995, 45, 328–336. Искать в Google Scholar
[93] Hong J, Tsao GT, Wankat PC. Biotechnol. Bioeng. 1981, 23, 1501–1516. Искать в Google Scholar
[94] Knutsen JS, Davis RH. Заявл. Biochem. Biotechnol. 2004, 113–116, 585–599. Искать в Google Scholar
[95] Mores WD, Knutsen JS, Davis RH. Заявл. Biochem.Biotechnol. 2001, 91–93, 297–309. Искать в Google Scholar
[96] Zhu Y, Malten M, Torry-Smith M, McMillan JD, Stickel JJ. Биоресурсы. Technol. 2011, 102, 2897–2903. Искать в Google Scholar
[97] Кристенсен Дж. Б., Фелби С., Йоргенсен Х. Прил. Biochem. Biotechnol. 2009, 156, 557–562. Искать в Google Scholar
[98] Рош К.М., Диббл С.Дж., Кнутсен Дж. С., Стикель Дж. Дж., Либераторе М. В.. Biotechnol. Bioeng. 2009, 104, 290–300. Искать в Google Scholar
[99] Mohanram S, Amat D, Choudhary J, Arora A, Nain L. Сустейн. Chem. Процесс. 2013, 1, 15–27. Искать в Google Scholar
[100] Ye Z, Zheng Y, Li B, Borrusch MS, Storms R, Walton JD. PLoS One 2014, 9, e109885. Искать в Google Scholar
[101] Viikari L, Alapuranen M, Puranen T, Vehmaanperä J, Siika-aho M. Adv. Biochem. Англ. Biotechnol. 2007, 108, 121–145. Искать в Google Scholar
[102] D’Amore T, Stewart GG. Enzyme Microb. Tech. 1987, 9, 322–330. Искать в Google Scholar
[103] Chu BCH, Lee H. Biotechnol. Adv. 2007, 25, 425–441. Искать в Google Scholar
[104] Jeffries TW. Curr. Opin. Biotechnol. 2006, 17, 320–326. Искать в Google Scholar
[105] Дьен Б.С., Котта М.А., Джеффрис Т.В. Заявл. Microbiol. Biotechnol. 2003, 63, 258–266. Искать в Google Scholar
[106] Баллестерос М., Олива Дж. М., Негр М. Дж., Мансанарес П., Баллестерос И. Process Biochem. 2004, 39, 1843–1848. Искать в Google Scholar
[107] Шоу А.Дж., Подкаминер К.К., Десаи С.Г., Бардсли Дж. С., Роджерс С. Р., Торн П. Г., Хогсетт Д. А., Линд Л. Р.. Proc. Natl. Акад. Sci. 2008, 105, 13769–13774. Искать в Google Scholar
[108] Карими К., Эмтиази Г., Тахерзаде М.Дж. Enzyme Microb. Tech. 2006, 40, 138–144. Искать в Google Scholar
[109] Kim TH, Taylor F, Hicks KB. Биоресурсы. Technol. 2008, 99, 5694–5702. Искать в Google Scholar
[110] Ko JK, Bak JS, Jung MW, Lee HJ, Choi I-G, Kim TH, Kim KH. Биоресурсы. Technol. 2009, 100, 4374–4380. Искать в Google Scholar
[111] Zhu S, Wu Y, Yu Z, Zhang X, Wang C, Yu F, Jin S, Zhao Y, Tu S, Xue Y. Биосист. Англ. 2005, 92, 229–235. Искать в Google Scholar
[112] Пименова Н.В., Хэнли Т.Р. Заявл. Biochem. Biotechnol. 2004, 113–116, 347–360. Искать в Google Scholar
[113] Stickel JJ, Knutsen JS, Liberatore MW, Luu W, Bousfield DW, Klingenberg DJ, Scott CT, Root TW, Ehrhardt MR, Monz TO. Rheol. Acta 2009, 48, 1005–1015. Искать в Google Scholar
[114] Knutsen JS, Liberatore MW. J. Rheol. 2009, 53, 877–892.Искать в Google Scholar
[115] Wiman M, Palmqvist B, Tornberg E, Lidén G. Biotechnol. Bioeng. 2011, 108, 1031–1041. Искать в Google Scholar
[116] Dasari RK, Berson RE. Заявл. Biochem. Biotechnol. 2007, 136–140, 289–299. Искать в Google Scholar
[117] Viamajala S, McMillan JD, Schell DJ, Elander RT. Биоресурсы. Technol. 2009, 100, 925–934. Искать в Google Scholar
[118] Беннингтон, CPJ, Kerekes RJ, Grace JR. Банка. J. Chem. Англ. 1990, 68, 748–757. Искать в Google Scholar
[119] Ehrhardt MR, Monz TO, Root TW, Connelly RK, Scott CT, Klingenberg DJ. Заявл. Biochem. Biotechnol. 2010, 160, 1102–1115. Искать в Google Scholar
[120] Rosgaard L, Andric P, Dam-Johansen K, Pedersen S, Meyer AS. Заявл. Biochem. Biotechnol. 2007, 143, 27–40. Искать в Google Scholar
[121] Knutsen JS, Liberatore MW. Energy Fuels 2010, 24, 6506–6512.Искать в Google Scholar
[122] Саманюк JR, Scott TC, Root TW, Klindenberg DJ. J. Rheol. 2012, 56, 649–665. Искать в Google Scholar
[123] Пименова Н.В., Хэнли Т.Р. Заявл. Biochem. Biotechnol. 2003, 105–108, 383–392. Искать в Google Scholar
[124] Palmqvist B, Lidén G. Biotechnol. Биотопливо 2012, 5, 57–66. Искать в Google Scholar
[125] Sakata M, Ooshima H, Harano Y. Biotechnol. Lett. 1985, 7, 689–694.Искать в Google Scholar
[126] Roche CM, Dibble CJ, Stickel JJ. Biotechnol. Биотопливо 2009, 2, 28–39. Искать в Google Scholar
[127] Dasari RK. Осахаривание с высоким содержанием твердых частиц и исследования вязкости в биореакторе со скребковой поверхностью. Кандидатская диссертация. 2004. Поиск в Google Scholar
[128] Mais U, Esteghlalian AR, Saddler JN. Заявл. Biochem. Biotechnol. 2002, 98–100, 463–472. Искать в Google Scholar
[129] Ingesson H, Zacchi G, Yang B, Esteghlalian AR, Saddler JN. J. Biotechnol. 2001, 88, 177–182. Искать в Google Scholar
[130] Samaniuk JR, Scott CT, Root TW, Klingenberg DJ. Биоресурсы. Technol. 2011, 102, 4489–4494. Искать в Google Scholar
[131] Palmqvist B, Wiman M, Lidén G. Biotechnol. Биотопливо 2011, 4, 10–18. Искать в Google Scholar
[132] Lenting HBM, Warmoeskerken MMCG. J. Biotechnol. 2001, 89, 217–226. Искать в Google Scholar
[133] Riedlberger P, Brüning S, Weuster-Botz D. Bioprocess Biosys. Англ. 2013, 36, 927–935. Искать в Google Scholar
[134] Аден А., Рут М., Ибсен К., Джечура Дж., Нивз К., Шихан Дж., Уоллес Б., Монтегю Л., Слейтон А., Лукас Дж. Лигноцеллюлозная биомасса для проектирования и экономики процесса этанола с использованием совместного использования текущий предварительный гидролиз разбавленной кислотой и ферментативный гидролиз кукурузной соломы. Технический отчет NREL / TP- 2002, 510-32438, NREL. Искать в Google Scholar
[135] Hoyer K, Galbe M, Zacchi G. J. Chem. Technol. Biotechnol. 2009, 84, 570–577. Искать в Google Scholar
[136] Riedlberger P, Weuster-Botz D. Bioresour. Technol. 2012, 106, 138–146. Искать в Google Scholar
[137] Solomon J, Elson TP, Nienow AW, Pace GW. Chem. Англ. Commun. 1981, 11, 143–164. Искать в Google Scholar
[138] Elson TP, Cheesman DJ, Nienow AW. Chem. Англ. Sci. 1986, 41, 2555–2562. Искать в Google Scholar
[139] Amanullah A, Hjorth SA, Nienow AW. Chem. Англ. Sci. 1998, 53, 455–469. Искать в Google Scholar
[140] Dasari RK, Dunaway K, Berson RE. Energy Fuels 2009, 23, 4 92–497. Искать в Google Scholar
[141] Jørgensen H, Vibe-Pedersen J, Larsen J, Felby C. Biotechnol. Bioeng. 2007, 96, 862–870. Искать в Google Scholar
Этанол из целлюлозы: общий обзор
Этанол из целлюлозы: общий обзорИндекс | Поиск | Дом | Оглавление
Барсук, П.С. 2002. Этанол из целлюлозы: общий обзор. п. 17–21. В: Дж. Яник и А. Випки (ред.), Тенденции в новых культурах и новых применениях. АШС Пресс, Александрия, Вирджиния.
P.C. Барсук
ВВЕДЕНИЕ
Использование этанола в качестве альтернативного моторного топлива постоянно растет. по всему миру по ряду причин. Внутреннее производство и использование этанола поскольку топливо может снизить зависимость от иностранной нефти, сократить торговый дефицит, создать рабочие места в сельской местности, сокращение загрязнения воздуха и уменьшение глобального изменения климата накопление углекислого газа.Этанол, в отличие от бензина, представляет собой кислородсодержащее топливо, которое содержит 35% кислорода, что снижает выбросы твердых частиц и NOx при сгорании.
Этанол может быть получен синтетическим путем из нефти или путем микробной конверсии. материалов биомассы путем ферментации. В 1995 г. около 93% этанола в мире произведено методом ферментации и около 7% синтетическим метод. Метод ферментации обычно состоит из трех этапов: (1) формирование раствора ферментируемых сахаров, (2) ферментация этих сахаров до этанол, и (3) разделение и очистка этанола, обычно путем перегонка.
САХАР КРЕМ
В ферментации участвуют микроорганизмы, которые используют сбраживаемый сахар в пищу. и в процессе производит этанол и другие побочные продукты. Эти микроорганизмы обычно можно использовать 6-углеродные сахара, одним из наиболее распространенных является глюкоза. Следовательно, материалы биомассы, содержащие высокие уровни глюкозы или прекурсоров в глюкозу легче всего преобразовать в этанол. Однако, поскольку сахарные материалы входят в пищевую цепочку человека, эти материалы обычно слишком дороги в использовании для производства этанола.
Одним из примеров сахарного сырья является сахарный тростник. Бразилия разработала успешный программа получения этанола из сахарного тростника по ряду причин: (1) Бразилия традиционно в значительной степени полагались на импортную нефть в качестве транспортного топлива, что вызвало серьезный экономическая утечка в стране; (2) Бразилия может добиться очень высоких урожаев сахарного тростника; и (3) Бразилия также переживала периоды плохих рынков сахара. Как результат, Правительство Бразилии разработало программы поддержки отрасли с результат того, что Бразилия смогла успешно производить и использовать сахарный тростник для производства топливного этанола.
Хотя грибы, бактерии и дрожжевые микроорганизмы могут использоваться для ферментации, специфические дрожжи ( Saccharomyces cerevisiae , также известные как Bakers ’ дрожжи, так как они обычно используются в хлебопекарной промышленности) часто используется сбраживать глюкозу до этанола. Теоретически 100 граммов глюкозы произведут 51,4 г этанола и 48,8 г диоксида углерода. Однако на практике микроорганизмы используйте немного глюкозы для роста, и фактический урожай будет менее 100%.
Другое сырье биомассы, богатое сахарами (материалами, известными как сахариды), включает: сахарная свекла, сладкое сорго и различные фрукты. Однако эти материалы все в пищевой цепи человека и, за исключением некоторых остатков обработки, обычно слишком дорого использовать для производства топливного этанола.
КРАСИВЫЕ СЫРЬКИ
Еще одно потенциальное сырье для этанола — крахмал. Молекулы крахмала состоят из длинных цепочек молекул глюкозы.Таким образом, крахмалистые материалы также можно ферментировать. после расщепления молекул крахмала на простые молекулы глюкозы. Примеры крахмалистых материалы, обычно используемые во всем мире для производства этанола, включают зерновые зерна, картофель, сладкий картофель и маниока. Зерновые, обычно используемые в США для производства этанола включают кукурузу и пшеницу.
В 1990 году в мире было произведено около 475 миллионов тонн кукурузы. около 200 миллионов тонн произведено в США.Примерно от 8 до 9 млн т, или 4% зерна кукурузы в США пошло на этанол в 1990 году. Бушель кукурузного зерна (25,3 кг или 56 фунтов при влажности 15%) может производить от 9,4 до 10,9 л (от 2,5 до 2,9 галлона) чистого этанола, в зависимости от используемой технологии.
Крахмалистые материалы требуют реакции крахмала с водой (гидролиз) до расщепить крахмал на сбраживаемый сахар (осахаривание). Обычно гидролиз осуществляется путем смешивания крахмала с водой с образованием суспензии, которая затем перемешивают и нагревают до разрыва клеточных стенок.Специфические ферменты, которые разрывает химические связи, добавляется в разное время во время нагрева цикл.
ЦЕЛЛЮЛОЗНЫЕ КОРМА
Подобно сахарным материалам, крахмалистые материалы также входят в пищевую цепочку человека и таким образом дороги. К счастью, существует третья альтернатива — целлюлозная материалы. Примерами целлюлозных материалов являются бумага, картон, дерево и другой волокнистый растительный материал.
Ресурсы целлюлозы в целом очень широко распространены и богаты.Например, леса составляют около 80% мировой биомассы. Изобилие и за пределами пищевая цепь человека делает целлюлозные материалы относительно недорогим сырьем для производства этанола.
Целлюлозные материалы состоят из лигнина, гемицеллюлозы и целлюлозы. и поэтому иногда их называют лигноцеллюлозными материалами. Один из основных Функции лигнина — обеспечивать структурную поддержку растения. Таким образом, в Как правило, в деревьях содержание лигнина выше, чем в травах.К сожалению, лигнин не содержит сахаров, содержит молекулы целлюлозы и гемицеллюлозы, что делает их труднодоступными.
Молекулы целлюлозы состоят из длинных цепочек молекул глюкозы, как и крахмал. молекулы, но имеют иную структурную конфигурацию. Эти структурные характеристики плюс инкапсуляция лигнином затрудняет целлюлозные материалы гидролизовать, чем крахмалистые материалы.
Гемицеллюлоза также состоит из длинных цепочек молекул сахара; но содержит, помимо глюкозы (6-углеродный или гексозный сахар) содержит пентозы (5-углеродный сахара).Чтобы усложнить ситуацию, точный сахарный состав гемицеллюлозы может варьироваться в зависимости от вида растения.
Поскольку 5-углеродные сахара составляют высокий процент доступных сахаров, способность восстанавливать и сбраживать их в этанол важна для эффективности и экономика процесса. В последнее время генетически были обнаружены особые микроорганизмы. разработан, который может сбраживать 5-углеродные сахара в этанол с относительно высокими эффективность.
Одним из примеров является генно-инженерный микроорганизм, разработанный университетом. Флориды, которая способна сбраживать как 5-, так и 6-углеродные сахара. Этот на микроорганизм был выдан патент США 5,000,000. Другие исследователи разработали микроорганизмы, способные эффективно ферментировать хотя бы часть присутствуют сахара.
Бактерии привлекли особое внимание исследователей из-за своей скорости брожения.Как правило, бактерии могут ферментироваться за считанные минуты по сравнению с часов для дрожжей.
ЭТАНОЛ ИЗ ЦЕЛЛЮЛОЗЫ
Во времена нехватки топлива ферментационный этанол производился в промышленных масштабах. в США из сырья целлюлозной биомассы с использованием методов кислотного гидролиза. В настоящее время в некоторых странах с более высокими ценами на этанол и топливо производят этанол из целлюлозного сырья. Однако это только недавно что рентабельные технологии производства этанола из целлюлозы (EFC) в США начали появляться.
Существует три основных типа процессов EFC — кислотный гидролиз, ферментативный. гидролизный и термохимический — с вариациями для каждого. Самый распространенный кислотный гидролиз. Можно использовать практически любую кислоту; однако серная кислота чаще всего используется, так как он обычно наименее дорогостоящий.
КИСЛОТНЫЙ ГИДРОЛИЗ
Существует два основных типа кислотных процессов: разбавленная кислота и концентрированная кислота, каждый с вариациями. Процессы с разбавленной кислотой проводятся при высоких температурах. и давление, и время реакции находится в диапазоне секунд или минут, что облегчает непрерывную обработку.
В качестве примера, используя процесс разбавленной кислоты с 1% -ной серной кислотой в непрерывном проточный реактор при времени пребывания 0,22 минуты и температуре 237 ° C (458 ° F) с чистой целлюлозой обеспечил выход сахаров более 50%. В этом случае, Из 0,9 т (1 тонны) сухой древесины можно получить около 189 л (50 галлонов) чистого этанола. Комбинация кислоты и высокой температуры и давления диктует особый реактор. материалы, которые могут сделать реактор дорогим.
Большинство процессов с разбавленной кислотой ограничиваются эффективностью извлечения сахара около 50%. Причина в том, что как минимум две реакции являются частью этого процесса. Первая реакция превращает целлюлозные материалы в сахар, а вторая — реакция превращает сахара в другие химические вещества. К сожалению, условия которые вызывают первую реакцию, также являются правильными условиями для второе, чтобы произойти. Таким образом, как только молекулы целлюлозы распадаются, реакция быстро расщепляет сахар на другие продукты, в первую очередь фурфурол, химическое вещество, используемое в пластмассовой промышленности.Не только разложение сахара снизить выход сахара, но фурфурол и другие продукты распада могут быть ядовитыми к микроорганизмам ферментации.
Самым большим преимуществом процессов с разбавленной кислотой является их высокая скорость реакции, что облегчает непрерывную обработку. Их самый большой недостаток — это их низкий выход сахара. Для быстрых непрерывных процессов, чтобы обеспечить адекватное проникновение кислоты, сырье также должно быть уменьшено в размере, чтобы максимально размер частиц находится в диапазоне нескольких миллиметров.
Поскольку 5-углеродные сахара разлагаются быстрее, чем 6-углеродные сахара, один способ Уменьшение деградации сахара должно проходить в два этапа. Первый этап — это проводится в мягких условиях процесса для извлечения 5-углеродных сахаров, в то время как второй этап проводится в более жестких условиях для извлечения 6-углеродного сахара. К сожалению, деградация сахара по-прежнему является проблемой, и урожайность ограничена. примерно до 272 л / т (80 галлонов этанола на тонну) сухой древесины.
Процесс концентрированной кислоты использует относительно мягкие температуры и единственный задействованные давления обычно возникают только при перекачивании материалов из судно к судну. Один процесс концентрированной кислоты был впервые разработан Министерством сельского хозяйства США. и далее уточняется Университетом Пердью и Управлением долины Теннесси.
В процессе производства концентрированной кислоты TVA кукурузная солома смешивается с разбавленным (10%) серной кислоты и нагревают до 100ºC в течение 2-6 часов в первом (или гемицеллюлозу) реактор гидролиза.Низкие температуры и давления сводят к минимуму деградацию сахаров. Для извлечения сахаров гидролизованный материал в первом реакторе замачивают в воде и несколько раз сливают.
Затем твердый остаток с первой стадии обезвоживают и замачивают в 30% -м растворе. до 40% концентрации серной кислоты в течение 1-4 часов в качестве предварительного гидролиза целлюлозы шаг. Затем этот материал обезвоживают и сушат, чтобы кислота концентрация в материале увеличивается примерно до 70%.После реакции в другом сосуд в течение 1-4 часов при 100ºC, содержимое реактора фильтруют для удаления твердые вещества и восстановить сахар и кислоту. Раствор сахара / кислоты из второго стадия рециркулируется на первую стадию, чтобы обеспечить кислоту для первой стадии гидролиз. Таким образом, сахара, полученные на второй стадии гидролиза, восстанавливаются в жидкость с первой ступени гидролиза.
Основным преимуществом концентрированного процесса является высокое извлечение сахара. эффективность, которая может составлять порядка 90% как гемицеллюлозы, так и целлюлозные сахара.Используемые низкие температуры и давления также позволяют использование относительно недорогих материалов, таких как резервуары из стекловолокна и трубопроводы. К несчастью, это относительно медленный процесс, и экономичные системы восстановления кислоты имеют было сложно развить. Без регенерации кислоты большие количества извести должны использоваться для нейтрализации кислоты в растворе сахара. Эта нейтрализация формирует большое количество сульфата кальция, который требует утилизации и создает дополнительные расход.
Используя некоторые предполагаемые коэффициенты конверсии целлюлозы и ферментации, этанол урожайность глюкозы может быть рассчитана для соломы кукурузной (надземная часть растения кукурузы за вычетом колосьев), как показано в таблице 1, показывающей выход этанола из глюкоза. Аналогичным образом можно рассчитать выход этанола из ксилозы, как показано на рисунке. в таблице 2.
Таблица 1. Выход этанола из глюкозы.
| Сухая солома | 1 тонна (1000 кг) |
| Содержание целлюлозы | 0.45 |
| Эффективность преобразования и извлечения целлюлозы | 0,76 |
| Стехиометрический выход этанола | 0,51 |
| Эффективность ферментации глюкозы | 0,75 |
| Выход глюкозы | 131 кг этанола = 151 л (40 галлонов) |
Таблица 2. Выход этанола из ксилозы.
| Сухая солома | 1 тонна (1000 кг) |
| Содержание гемицеллюлозы | 0.29 |
| Эффективность преобразования и извлечения гемицеллюлозы | 0,90 |
| Стехиометрический выход этанола | 0,51 |
| Эффективность ферментации ксилозы | 0,50 |
| Выход ксилозы | 66 кг этанола = 76 л (20 галлонов) |
Таким образом, в этом примере общий выход на тонну сухой соломки составляет около 227 л (60 галлонов) этанола.Эти числа также показывают, насколько важны конверсия сахара и эффективности восстановления и эффективности ферментации. Если бы можно было достичь 95% для обоих коэффициентов полезного действия, тогда выход будет примерно 350 л / т (103 галлонов этанола / тонна).
ЭНЗИМАТИЧЕСКИЙ ГИДРОЛИЗ
Другой основной метод гидролиза — ферментативный гидролиз. Ферменты естественно встречающиеся растительные белки, которые вызывают определенные химические реакции. Тем не мение, чтобы ферменты работали, они должны получить доступ к молекулам, подлежащим гидролизу.Чтобы ферментативные процессы были эффективными, требуется предварительная обработка. таким образом необходимо разрушить кристаллическую структуру лигноцеллюлозы и удалить лигнин, чтобы обнажить молекулы целлюлозы и гемицеллюлозы. В зависимости от материал биомассы, физические или химические методы предварительной обработки могут быть использовал.
Физические методы могут использовать высокую температуру и давление, измельчение, облучение, или замораживание — все это требует большого расхода энергии.Химический Метод использует растворитель для разрушения и растворения кристаллической структуры.
Пример процесса на основе ферментативного гидролиза разрабатывается Национальная лаборатория возобновляемых источников энергии (NREL). После предварительной обработки разбавленной кислотой, суспензия очищается от токсинов, чтобы удалить материалы, которые могут быть ядовиты для микроорганизмы, используемые в процессе. Небольшая часть этого шлама отправляется в отдельный сосуд, который используется для выращивания микроорганизмов, производящих целлюлазу фермент для процесса.Другая часть пульпы отправляется в другую емкость. для поддержания и выращивания дрожжевой культуры для брожения. В процессе NREL, и ферменты, и ферментационные микроорганизмы добавляются одновременно в суспензию, а конверсия сахара и ферментация происходят одновременно в процесс, называемый одновременным осахариванием и коферментацией (SSCF).
Из-за прочной кристаллической структуры имеющиеся в настоящее время ферменты требуют несколько дней для достижения хороших результатов.Поскольку длительное время технологического процесса связывает реактор судов в течение длительного времени, эти суда должны быть либо достаточно большими, либо иметь много из них необходимо использовать. Любой вариант стоит дорого. В настоящее время стоимость ферментов также слишком высока, и исследования продолжаются, чтобы снизить стоимость ферментов.
Однако, если будут разработаны менее дорогие ферменты, ферментативные процессы сохранятся. несколько преимуществ: (1) их эффективность достаточно высока, а их побочные продукты можно контролировать; (2) их мягкие технологические условия не требуют дорогостоящих материалы конструкции; и (3) их технологические потребности в энергии относительно низкий.
ТЕРМОХИМИЧЕСКИЕ ПРОЦЕССЫ
Существует два процесса производства этанола, в которых в настоящее время используются термохимические реакции в их процессах. Первая система на самом деле является гибридной термохимической. и биологическая система. Примером может служить процесс, разрабатываемый компанией Bioengineering. Ресурсы в Фейетвилле, Арканзас. Материалы биомассы являются первыми термохимическими газифицируется и синтез-газ (смесь водорода и оксидов углерода) барботируется через специально сконструированные ферментеры.Микроорганизм, способный преобразовывать синтез-газ вводится в ферментеры при определенных условиях процесса вызвать брожение до этанола.
Второй процесс термохимического производства этанола не использует никаких микроорганизмов. В этом процессе материалы биомассы сначала термохимически газифицируются, и синтез-газ проходит через реактор, содержащий катализаторы, которые вызывают газ превращается в этанол.Германия предприняла активные усилия в Вторая мировая война разработала эти процессы для топлива. Были предприняты многочисленные усилия. сделано с тех пор для разработки коммерчески жизнеспособных термохимических процессов получения этанола.
Достигнут выход этанола до 50% с использованием синтез-газа в этанол. процессы. Некоторые процессы, которые сначала производят метанол, а затем используют каталитические сдвиги по производству этанола дали выход этанола в диапазоне 80%. К сожалению, как и в случае с другими процессами, поиск рентабельного полностью термохимического процесс был трудным.
УСИЛИЯ ПО КОММЕРЦИАЛИЗАЦИИ
Несколько заводов EFC были построены и эксплуатировались в разных странах во время мировой войны. II, когда условия военного времени изменили экономические условия и приоритеты. Эти страны включали Германию, Россию, Китай, Корею, Швейцарию, США, и другие страны. Сегодня из-за конкуренции со стороны синтетического этанола, только несколько из этих заводов все еще работают, практически все они в Россия.
Завод по производству бумаги в Теми-Шаммайге, Квебек, работает за счет побочных продуктов. сахара, содержащиеся в «сульфитном растворе», который содержит около 2% сбраживаемых сахара. Это единственный в своем роде объект в Северной Америке. Этот объект управляется Tembec, Inc., и производит 4 миллиона галлонов промышленной продукции в год. этанол марки.
В Северной Америке предпринимаются усилия по коммерческому производству этанола. из дерева и других целлюлозных материалов в качестве основного продукта.Таблица 3 частично резюмирует эти компании и их деятельность, которые находятся в разных состояниях. прогресса.
Таблица 3. Частичный список компаний, разрабатывающих этанол из целлюлозы. технологии.
| Расположение компании и головного офиса | Технологии | Первичное сырье | Емкость по этанолу | Комментарии |
| BCI, Дедхэм, Массачусетс | Разбавленная кислота | Багасса | 7560 миллионов л / год (20 миллионов галлонов в год z ) | Завод откроется в 2002 году |
| Bioengineering Resources, Fayetteville, AR | Термохимическая газификация с ферментацией | Опытно-промышленная установка | ||
| Ethxx International, Аврора, ON | Термохимическая газификация с каталитической конверсией | Дерево | Опытно-промышленная установка | |
| Fuel Cell Energy, Lakewood, CO | Термохимическая газификация с каталитической конверсией | Дерево | Опытно-промышленная установка | |
| Иоген, Оттава, ON | Ферментативный | Овсяная шелуха, просо, пшеничная солома и кукурузная солома | 378 миллионов л / год (1 миллион галлонов в год) | Экспериментальный завод действующий |
| Масада, Бирмингем, AL | Кислота концентрированная | MSW | 3780 миллионов л / год (10 миллионов галлонов в год) | Завод в начале 2002 г. |
| Paszner Technologies, Inc, Суррей, Британская Колумбия | Подкисленный водно-ацетоновый процесс | Дерево | Строящиеся коммерческие предприятия | |
| PureVision Technology, Ft.Lupton, CO | Ферментативный | Дерево | Строительство опытного завода |
РЕЗЮМЕ
Этанол из целлюлозы (EFC) имеет большой потенциал благодаря широкой доступности, обилие и относительно невысокая стоимость целлюлозных материалов. Однако хотя несколько процессов EFC технически осуществимы, рентабельные процессы имеют было трудно достичь.Только недавно появились экономичные технологии EFC начали появляться.
Unasylva — № 125 — Химия из дерева
Unasylva — № 125 — Химия из дерева — Химия из дереваПерспективы на будущее
Ирвинг С. Голдштейн
ИРВИНГ, С. ГОЛДШТЕЙН — профессор кафедры древесины и бумаги Государственного университета Северной Каролины, США.Эта статья представляла собой документ, который был представлен на Восьмом Всемирном лесном конгрессе, Джакарта, октябрь 1978 г.
Древесина в химической промышленности имеет два будущих применения. Один заключается в расширении и расширении существующих практик. Другой — преобразование компонентов клеточной стенки в химическое сырье. Эти полимеры клеточной стенки, составляющие основную часть древесины, могут удовлетворить все наши химические потребности вместо нефтехимии.
Прогнозы будущего спроса на некоторые товары могут основываться на росте населения или прошлых тенденциях.Однако при прогнозировании будущего спроса на химические вещества из древесины простая экстраполяция имеющихся данных не является надежной. В настоящее время индустриальные общества почти полностью зависят от нефтехимических продуктов, полученных из ископаемых жидких и газообразных углеводородов, в качестве сырья для волокон, пластмасс, каучуков, клеев и т. Д. Поскольку нефть и природный газ становятся более дорогими и дефицитными, очевидно, что другие источники углерода, такие как уголь или древесина, могут стать альтернативным сырьем. Однако, несмотря на то, что переработка древесины в химические вещества является концептуально и технологически осуществимой, из этого не обязательно следует, что древесина будет использоваться для этой цели.
Прежде чем исследовать факторы — которые в основном не поддаются контролю лесной промышленности — которые в конечном итоге определят вид и объем химических веществ, которые будут производиться из древесины в будущем, было бы полезно понять потенциальный вклад древесины в эта зона. Этот фон представлен здесь в виде краткого обсуждения химического состава древесины и краткого описания прошлых, настоящих и потенциальных будущих химических веществ, полученных из древесины.
Хотя теоретически химик может преобразовать компоненты древесины в любые желаемые химические вещества, химические вещества, которые на практике могут быть получены из древесины, в значительной степени зависят от присущего ей химического состава.Древесина представляет собой смесь трех природных полимеров — целлюлозы, лигнина и гемицеллюлозы — в примерном соотношении 50:25:25, в зависимости от вида, биологических вариаций, таких как генетические различия внутри видов, и условий выращивания. Целлюлоза и гемицеллюлозы представляют собой углеводные полимеры, состоящие из молекул простых сахаров, а лигнин представляет собой полимер фенилпропановых звеньев (Browing, 1963).
Целлюлоза представляет собой длинноцепочечный полимер глюкозы, который отличается от крахмала, также полимера глюкозы, только конфигурацией молекул глюкозы.Волокнистая природа древесных клеток является результатом линейного, ориентированного, кристаллического расположения их самого распространенного компонента, целлюлозы.
Гемицеллюлозы — это более короткие или разветвленные полимеры пятиуглеродных сахаров (пентозы), таких как ксилоза, или шестиуглеродных сахаров (гексозы), кроме глюкозы. Они аморфны по своей природе и вместе с лигнином образуют матрицу, в которую встроены фибриллы целлюлозы. Хотя структура целлюлозы одинакова у разных пород, гемицеллюлозы значительно различаются между видами, и особенно между лиственной и мягкой древесиной.Гемицеллюлозы лиственных пород обычно богаче пентозами, тогда как гемицеллюлозы хвойных пород обычно содержат больше гексоз.
Лигнин, третий компонент клеточной стенки, представляет собой трехмерный полимер, образованный из фенилпропановых звеньев, которые случайным образом выросли в сложную большую молекулу с множеством различных видов связей между строительными блоками. Структура лигнина также различается между лиственными и хвойными породами. Фенильные группы в лигнинах древесины твердых пород более сильно замещены метоксильными группами, чем в лигнинах древесины мягких пород.Одним из следствий этого различия является то, что лигнины древесины твердых пород менее сшиты и легче растворяются при варке целлюлозы.
ЛЕС В КАНАДЕ химический завод
Лигнин действует как цемент между древесными волокнами и как агент, придающий жесткость волокнам. При производстве химической древесной массы она растворяется в различных химических процессах, оставляя целлюлозу и гемицеллюлозу в волокнистой форме. Некоторые гемицеллюлозы теряются в процессе из-за их более низкой молекулярной массы, большей растворимости и более легкого гидролиза.
В дополнение к полимерным компонентам клеточной стенки, которые составляют основную часть древесины, различные породы содержат различные количества и виды посторонних материалов, вместе называемых экстрактивными веществами. В древесине хвойных пород мы часто находим значительные количества смол, состоящих как из жирных кислот, так и из так называемых смоляных кислот, которые являются производными простых терпенов, таких как скипидар и сосновое масло. Танины — это полигидроксильные фенолы, которые содержатся в сердцевине древесины и коре многих видов.Каучук получают из внутренней коры некоторых деревьев в виде латекса. Ароматические масла и водорастворимые сахара также могут быть получены из различных видов. Знакомые примеры — кедровое масло и кленовый сахар.
ПИРОЛИЗ ДРЕВЕСИНЫ. До появления дешевого ископаемого топлива, такого как уголь, нефть и природный газ, деструктивная перегонка древесины для производства древесного угля была важной отраслью промышленности (Stamm, 1953). В Соединенных Штатах остаются только остатки отрасли, производящей брикеты для приготовления пищи на открытом воздухе.
Газ, образующийся при карбонизации древесины, можно использовать в качестве топлива с низким содержанием БТЕ. Иногда он использовался во время Второй мировой войны для питания двигателей внутреннего сгорания, когда бензин был недоступен. Ряд летучих органических химикатов можно извлечь из дистиллята пиролиза древесины. Уксусная кислота, метиловый спирт и ацетон раньше получали исключительно путем перегонки древесины, что привело к общепринятому названию «древесный спирт» для метанола. Кроме того, были выделены различные фракции древесного дегтярного масла, используемые в медицине, копчении мяса, дезинфицирующих средствах и средствах для уничтожения сорняков.
ЭКСТРАКТИВЫ. Смолистые экссудаты сосен стали сырьем для военно-морских магазинов, старейшей химической промышленности в Северной Америке. Эти неочищенные экссудаты, которые сначала использовались для смолки канатов и заделки швов на деревянных кораблях, затем перегоняли для получения скипидара и канифоли, которые находят широкое применение в промышленности.
Латекс, полученный из каучуковых деревьев, в течение многих лет был единственным источником каучука. Для многих применений натуральный каучук по-прежнему предпочтительнее синтетического каучука для плитки.Экстракты сердцевины некоторых твердых пород древесины, а также коры различных пород обеспечивали дубильные вещества, которые, как следует из их названия, играли важную роль при дублении кожи.
В дополнение к целлюлозе, наиболее широко используемому сегодня полимеру, который в основном используется в его естественном волокнистом состоянии после извлечения из древесины, значительные количества химикатов, называемых «сильвихимикатами», все еще используются сегодня (Goheen, 1972), несмотря на подавляющее преобладание. химических веществ на нефтяной основе, называемых «нефтехимия».»
ЖИВОТЫ ДЛЯ ПУЛЬПИРОВАНИЯ. Химические фрагменты полимеров клеточной стенки, которые попадают в раствор после варки целлюлозы, часто можно выделить из щелоков варки и использовать. Как правило, щелочные растворы для варки целлюлозы сжигают при регенерации химикатов для варки целлюлозы, но сульфитные щелоки для варки целлюлозы часто обрабатывают для получения полезных побочных продуктов.
Сульфированный лигнин можно осаждать в виде сульфонатов лигнина и использовать в качестве дубильных агентов, клеев, связующих, диспергаторов и т. Д.Сахар в отработанном сульфитном щелоке можно сбраживать дрожжами с получением этилового спирта, кормов и пищевых добавок. Путем мягкого щелочного окисления сульфонатов лигнина можно получить ванилин для ароматизаторов и отдушек.
Щелочной лигнин из сульфата или крафт-черного щелока может осаждаться и использоваться в качестве наполнителя для смол, для армирования резины и стабилизации эмульсии. Летучие продукты из крафт-черного щелока включают диметилсульфид, диметилсульфоксид и диметилсульфон, используемые в качестве растворителей и химических реагентов.
НЕФТЬ. Промышленность по производству жевательной резинки для кораблей по большей части была заменена извлечением масляно-смоляных компонентов древесины из процесса варки крафт-целлюлозы (Uprichard, 1978; Zinkel, 1975). Летучие вещества, такие как скипидар, извлекаются из выпускных газов варочного котла. Щелочной раствор для варки целлюлозы превращает жирные кислоты и смоляные кислоты в соли натрия, которые можно удалить из концентрированного черного щелока и подкисить с получением сырого таллового масла.
ГИДРОЛИЗ ДЕРЕВА. Гидролиз древесины, превращение углеводных полимеров в древесине в простые сахара путем химической реакции с водой в присутствии кислотных катализаторов, известен уже 150 лет и практиковался в промышленных масштабах в Соединенных Штатах во время Первой мировой войны. Германия во время Второй мировой войны, а сейчас в СССР.Глюкоза является основным продуктом и может быть далее преобразована в этанол или дрожжи.
ЦЕЛЛЮЛОЗНЫЕ ПОЛИМЕРЫ. Химическая целлюлоза высокой чистоты или растворяющаяся пульпа является исходным материалом для таких производных полимерной целлюлозы, как вискоза и целлофан (регенерированная целлюлоза), сложные эфиры целлюлозы, такие как ацетат и бутират для волокон, пленки и формования, а также простые эфиры целлюлозы, такие как карбоксиметилцеллюлоза, этилцеллюлоза и гидроксиэтилцеллюлоза для использования в качестве камедей.
ЭКСТРАКТИВЫ.Некоторое количество скипидара и канифоли до сих пор получают путем паровой дистилляции или экстракции сосновых пней. Арабиногалактан, гемицеллюлозная камедь, извлеченная из лиственницы, используется вместо пахотной камеди. Фенольные кислоты, извлеченные из коры различных хвойных деревьев, используются в качестве наполнителей для клеев на основе синтетических смол, а также в качестве связующих и диспергаторов. Воски, извлеченные из коры пихты Дугласа, могут использоваться для общих восковых применений, а натуральный каучук остается важным материалом: материалом.
Будущее использование древесины для производства химикатов можно разделить на две основные категории, первая из которых в основном связана с расширением и расширением существующей практики, а вторая связана с преобразованием полимеров клеточной стенки в химическое сырье с низкой молекулярной массой.
РАСШИРЕНИЕ СОВРЕМЕННОЙ ПРАКТИКИ. Здесь речь идет не только о физическом расширении существующих операций, но и о расширении соответствующих возможностей в использовании побочных продуктов и добывающих материалов. Приведенные примеры являются иллюстративными, а не ограничивающими.
Экстракты коры и древесины обладают гораздо большим потенциалом, чем их нынешнее использование (Hillis, 1978; Laver, 1978). Целлюлозные полимеры могут стать более важными, если можно будет снизить затраты на энергию и улучшить свойства (Allen, 1978; Goldstein, 1977).Выработку олеорезина в соснах можно стимулировать применением гербицидов (Робертс, 1973). Углеводородные полимеры можно получить при выращивании новых растений в качестве товарных культур (Galvin, 1978). Фенолы с низкой молекулярной массой могут быть получены из побочного продукта лигнина в варочных растворах (Goheen, 1971; Benigni and Goldstein, 1971). Сахариновые кислоты могут быть извлечены из крафт-черного щелока (Sarkanen, 1976). Листва может давать эфирные масла, хлорофилл и белок (Barton, 1978).
ПРЕОБРАЗОВАНИЕ ПОЛИМЕРОВ КЛЕТОЧНЫХ СТЕН.Основная часть древесины состоит из компонентов клеточной стенки. Этот источник сырья по объему намного превышает экстрактивные компоненты или химические побочные продукты и представляет собой потенциальный ресурс для удовлетворения всех наших химических потребностей вместо нефтехимии. Крупномасштабное производство промышленно важных химикатов из лигноцеллюлозы может осуществляться различными способами (Goldstein, 1976a). Эти тоннажи могут обеспечить основные химические строительные блоки для преобразования в синтетические полимеры (Goldstein, 1975a).
ВСЕГО ДЕРЕВА. Полимеры клеточной стенки в их естественном смешанном состоянии могут быть разбиты на более простые соединения радикальными процессами, включающими высокие температуры, а в некоторых случаях и высокое давление. Эти процессы по своей природе неселективны и аналогичны тем, которые используются для преобразования твердых органических отходов или угля в химические вещества.
При газификации древесина нагревается до температуры 1 000 ° C с образованием смеси оксида углерода и водорода в качестве основных продуктов (Prahacs, 1971).В качестве побочных продуктов образуются этилен, ацетилен, пропилен, бензол и толуол с низким выходом. Монооксид углерода и водород, образующиеся при газификации угля, могут быть (а) дополнительно обработаны для получения водорода для производства аммиака, (б) каталитически преобразованы в метанол, (в) обогащены водородом и прореагированы с образованием метана, или (г) каталитически преобразованы к смеси алифатических углеводородов по процессу Фишера-Тропша.
Древесина может быть сжижена реакцией с оксидом углерода и водой при 350400 ° C и давлении 4 000 фунт-сила / дюйм² в присутствии различных катализаторов (Appel, 1971).Вязкая нефть производится с выходом 40–50%, которая может быть переработана в химические вещества так же, как нефтехимические продукты получают из нефти.
Пиролиз или термическое разложение древесины в отсутствие воздуха или кислорода превращает древесину в древесный уголь, газ и масло (Soltes, 1978; Wender, 1974). Относительный выход продуктов зависит от условий пиролиза и состава субстрата, но типичные значения при 900 ° C могут быть: 25-35 процентов древесного угля, 30-45 процентов газа и до 8 процентов гудрона и масла.Газ состоит в основном из водорода, окиси углерода и метана, а гудрон и нефть содержат такие легкие нефтяные компоненты, как бензол и толуол, а также смеси более высококипящих соединений.
ЦЕЛЛЮЛОЗА. Селективное превращение полимерной целлюлозы глюкозы в мономерную глюкозу можно осуществить несколькими способами. Гидролиз до глюкозы может катализироваться кислотами или ферментами, но ни один из этих способов не столь прост, как гидролиз крахмала, из-за кристаллической природы целлюлозы.Кислотный гидролиз не ингибируется содержанием лигнина, но целлюлоза с высоким содержанием лигнина устойчива к ферментативному гидролизу, поэтому древесина должна быть частично делигнифицирована или тонко измельчена, чтобы подвергнуться гидролизу ферментами.
Уголь или древесина станут альтернативой нефти как химическому сырью будущего. Это, конечно, будет зависеть от того, какая из них есть в стране. Но возобновляемой может быть только древесина.
Кислотный гидролиз разбавленной кислотой при высоких температурах вызывает разложение части образовавшейся глюкозы до гидроксиметилфурфурола (Harris, 1975), ограничивая чистый выход сахара примерно до 50 процентов.Сильный кислотный гидролиз при более низких температурах может обеспечить почти количественный выход глюкозы (Kusama, 1960).
Шафизаде (1978) показал, что сухая перегонка целлюлозы при 400–500 ° C дает около 80 процентов смолы, которая содержит в основном левоглюкозан и может быть преобразована в глюкозу с выходом 50 на
процента в расчете на целлюлозу. Целлюлоза, свободная от других компонентов клеточной стенки, потребуется, чтобы избежать загрязнения и реакции с другими продуктами разложения.
Превращение целлюлозы в глюкозу является первым шагом в потенциально широкомасштабном химическом использовании целлюлозы.Наибольшее потенциальное значение имеет ферментация глюкозы в этанол с помощью коммерчески проверенных методов с высокими выходами. Этанол является важным промышленным химическим веществом, которое в настоящее время производится путем гидратации этилена. Также он может найти широкое применение в качестве топлива для двигателей внутреннего сгорания.
Дегидратация этанола до этилена, обратная реакция для получения этанола из этилена из нефти, также приводит к высоким выходам. Точно так же бутадиен можно легко получить из этанола с помощью способов, которые были коммерчески доказаны, но стали устаревшими из-за дешевой нефти.Превращение глюкозы через этанол в этилен и бутадиен представляет собой наибольший потенциал использования целлюлозы для химических веществ из-за важности этилена как самого крупного органического химического вещества и как строительного блока для нефтехимии и пластмасс, а также бутадиена как агента в производство синтетического каучука.
Глюкоза также может быть преобразована в химические вещества, которые в настоящее время не имеют промышленного значения, но которые могут стать таковыми при надлежащих экономических условиях.Одним из таких процессов является производство гидроксиметилфурфурола и его дальнейшее превращение в левулиновую кислоту под действием горячих минеральных кислот на глюкозу (Harris, 1975). Использование глюкозы в качестве основного субстрата для ферментации позволит производить широкий ассортимент антибиотиков, химикатов, витаминов и ферментов из древесины (Seeley, 1976). Молочная кислота, например, может быть преобразована в акриловую кислоту и акрилаты.
ГЕМИЦЕЛЛЮЛОЗЫ. Гемицеллюлозы легче гидролизуются кислотой, чем целлюлоза, и, следовательно, легко превращаются в простые сахара в мягких условиях с высоким выходом.Ксиланы, которых больше в лиственных деревьях, дают в основном ксилозу, в то время как глюкоманнаны, содержащиеся в больших количествах в хвойных деревьях, дают маннозу.
Манноза и другие гексозы могут быть объединены с глюкозой для ферментации до этанола. Ксилозу и другие пентозы можно превратить в фурфурол или, альтернативно, ксилозу можно восстановить до ксилита. В настоящее время фурфурол коммерчески производится путем кислотной обработки ксилана в кукурузных початках и жоме сахарного тростника. Поскольку его можно будет получить в гораздо большем количестве при гидролизе древесины, потребуются новые применения.Однако, поскольку фурфурол когда-то служил сырьем для нейлона, пока его не вытеснил бутадиен, изобилие фурфурола по разумной цене должно стимулировать новые применения.
ЛИГНИН. Особенностями лигнина, важными для химического использования, являются его ароматический характер (в химическом смысле содержания фенильных групп) и ковалентная углерод-углеродная связь, которая предотвращает превращение в мономеры при мягкой обработке. Сообщается, что при более жестких условиях пиролиза, гидрирования и гидролиза выход фенолов достигает 50 процентов (Goheen, 1971, Goldstein, 1975b; Schweers, 1978), и, кроме того, прогнозируемые выходы фенола с чистотой 35 процентов имеют было предложено.Бензол был выделен как компонент при гидрокрекинге лигнина, и его можно было получить с выходом 25% по лигнину дегидроксилированием фенола.
БУДУЩИЙ ХИМИЧЕСКИЙ ЗАВОД. Из предыдущего обсуждения очевидно, что различные компоненты древесины могут быть преобразованы в определенные химические вещества: целлюлозу, например, в этанол, а гемицеллюлозы в фурфурол. Хотя время от времени рассматривались такие монопродуктовые заводы, они не представляют собой наиболее эффективное и экономичное использование древесины, заготовленной и собранной в качестве сырья.Лесохимический завод будущего должен преобразовывать все компоненты древесины в полезные продукты, так же как нефтеперерабатывающие заводы и мясокомбинаты используют все свое сырье.
Пример такой схемы (Goldstein, 1975a), но не единственный, который можно представить, будет включать предварительный гидролиз древесины слабыми кислотами для превращения гемицеллюлоз в потоки, богатые ксилозой или маннозой, с оставлением твердого остатка целлюлоза и лигнин сзади. Ксилозу можно превратить в фурфурол или ксилит, а маннозу объединить с глюкозой для ферментации.Сильный кислотный гидролиз целлюлозно-лигнинного субстрата дает раствор глюкозы, сбраживаемый до этанола или других химических веществ, и твердый остаток лигнина. Затем лигнин можно гидрогенизировать до фенолов или других обработанных смол. Путем дальнейшей переработки этанола в этилен и бутадиен такая установка могла бы производить химические вещества, необходимые для производства литья синтетических пластмасс, волокон и каучуков, необходимых промышленному обществу.
Могут быть разработаны и другие интегрированные системы использования древесины, которые включают альтернативное производство энергии, продуктов питания и материалов, а также химикатов (Липинский, 1978).Эти системы могут иметь более или менее химический компонент и в этой степени способствовать химическим потребностям людей во всем мире.
Древесина для производства химикатов не конкурирует с древесиной для производства деревянных панелей или целлюлозы. Форма не имеет значения. Вид и размер не имеют значения.
Существенная замена используемых в настоящее время нефтехимических продуктов химическими веществами из древесины будет зависеть от ряда технических, экономических, политических и социальных факторов. Концептуальная осуществимость химических веществ из древесины, разработанная в предыдущих разделах, будет зависеть от реальности факторов, обсуждаемых ниже.
НОВАЯ ЭРА. Химическая промышленность за последние 50 лет быстро выросла как в объеме, так и в плане качества продукции. Стимулируемые обильными и недорогими поставками нефти в качестве сырья, синтетические органические материалы стали настолько распространенными, что уровень жизни индустриальных обществ неразрывно связан с этими материалами.
Дэвис (1978) указал, что за эти 50 лет промышленная химия знала три эпохи. В первом случае впереди была свободная наука.Результаты хороших и актуальных исследований привели к изобретениям, которые оказались полезными и прибыльными. Во вторую эпоху после Второй мировой войны рыночные силы задавали темп. Выявление рыночных потребностей позволило изобретательности и науке удовлетворить их по-новому. Именно в эту эпоху дешевая нефть способствовала быстрому росту. Третью эпоху, в которую мы сейчас вступаем, Дэвис назвал «периодом ресурсов и земледелия». Теперь необходимо улучшить, удешевить и обеспечить поставки желаемой продукции за счет экономии материалов и замены более обильного сырья.
Согласно этому анализу, химическая промышленность должна обратиться к ресурсной базе, чтобы продолжать процветать. Точно так же, как раньше был переход от каменноугольной смолы к нефтяной, теперь для будущего химического сырья может потребоваться переход на уголь и целлюлозу. Химические вещества из древесины, вероятно, не будут сталкиваться с институциональным барьером для их принятия. Казалось бы, историческая адаптивность химической промышленности создает благоприятный климат.
УЛУЧШЕННАЯ ТЕХНОЛОГИЯ.Время и скорость, с которой химические вещества из древесины могут вытеснить нефтехимические продукты, в конечном итоге будут зависеть от их относительной стоимости. Цена на нефть не зависит от лесной промышленности, но изменение технологий в химии древесины может способствовать снижению этих затрат.
При нынешних ценах химические вещества из древесины, такие как этанол, метанол и т. Д., Не могут конкурировать с продуктами нефтехимии. Единственным возможным исключением может быть интегрированный завод по производству этанола, фурфурола и фенола (Katzen, 1978; USDA Forest Service, 1976).Улучшение процессов газификации древесины и гидролиза древесины с целью повышения урожайности и снижения капитальных затрат могло бы уменьшить этот ценовой разрыв, особенно если бы произошли значительные улучшения по сравнению с 50-процентным чистым выходом глюкозы при гидролизе слабой кислотой.
Выделение химической целлюлозы (растворяющаяся пульпа) и ее преобразование в изготовленные волокна, пленки, пластмассы или камеди — это области, в которых технологические усовершенствования могут привести к улучшению конкурентных позиций целлюлозных полимеров, не только по отношению к синтетическим полимерам из нефти, но также конкурируют с синтетическими полимерами из сырья, полученного при разложении целлюлозы.
Другими областями, в которых улучшенная технология может благоприятно повлиять на стоимость химических веществ из древесины, являются гидролиз целлюлозы до глюкозы ферментами и превращение лигнина в фенолы.
Исследования по химическому использованию древесины были относительно скудными по сравнению с усилиями, затраченными на нефтехимию за последние 50 лет. В процентах от продаж они составляют лишь около 10 процентов от общего объема продаж химикатов и синтетических полимеров.Фактически, когда цены на нефть падали, исследования химических веществ из древесины были практически прекращены. Можно ожидать, что увеличение масштабов исследований по древесине приведет к появлению значительных новых и улучшенных технологий, позволяющих полностью использовать потенциал древесины в качестве сырья для химических веществ.
Этанол может использоваться как моторное топливо, как промышленный химикат сам по себе и как сырье для производства этилена. Рынок для каждого разный, но цена на этанол, вероятно, будет определяться рынком для его использования в качестве топлива.
СТОИМОСТЬ И НАЛИЧИЕ. Этот фактор является наиболее важным фактором, влияющим на химические вещества из древесины, и одним из самых непредсказуемых. Реакция на рост стоимости нефти и признание ее окончательного истощения стимулировали проведение многих анализов альтернативных источников энергии и альтернативного природного химического сырья, еще одним примером которых является данная статья. Среди экспертов существуют большие разногласия по поводу того, насколько быстро цены на нефть будут продолжать расти и как скоро запасы будут исчерпаны.Эти вопросы неподконтрольны лесной промышленности. Сторонники древесины как альтернативного химического сырья пока ждут, мало что могут сделать, кроме улучшения своей технологии.
На недавней конференции по альтернативам химического сырья (Van Antwerper, 1977) одна группа участников предсказала, что нефтехимическая промышленность начнет падать примерно в 1990 году, при этом 50 или более процентов нового сырья будет добавлено в систему из других источников. -источники нефти. На другом конце шкалы прогнозов середина следующего столетия была названа приблизительной датой истощения мировых запасов нефти.Где-то в промежуточный период цены на сырую нефть будут настолько высокими, что производство этилена путем дегидратации этанола, вероятно, станет экономически целесообразным (Sarkanen, 1976). Это, конечно, предполагает, что цены на сырую нефть будут продолжать расти более быстрыми темпами, чем цены на древесину.
Некоторые люди утверждают, что нефть более ценна как химическое сырье, чем как топливо, и поэтому ее следует сохранять для преимущественного использования в качестве химического сырья. Однако, если пользователям нефтяного сырья придется конкурировать с потребителями энергии, те, кто потребует бензин и топочный мазут, будут готовы платить больше, тем самым улучшая конкурентные позиции альтернативного сырья.
УГОЛЬ И ДРЕВЕСИНА. В качестве альтернативы химическое сырье, получаемое в настоящее время из нефти, может быть получено из угля или древесины. Уголь также является ограниченным ресурсом и в конечном итоге будет потребляться, а в качестве возобновляемого ресурса навсегда останется только древесина. Но в ближайшем будущем уголь, вероятно, будет играть важную роль в химической промышленности, потому что разработки продвинулись дальше, чем с древесиной, и потому что массовое использование угля для производства синтетического жидкого топлива также приведет к образованию химикатов. Фактически, сейчас действуют установки газификации угля, особенно в Южной Африке.
Фактическая цена на такие химические вещества из угля будет зависеть от того, были ли они оценены по выгодной цене как побочные продукты или отражали справедливую долю затрат на производство. Инвестиционные затраты на установку для угля намного выше, чем для нефти, равно как и затраты на добычу и транспортировку. Стоимость угля сама по себе не отражала ущерба окружающей среде, так же как стоимость нефти до 1973 года не отражала истощение ресурса. Было предсказано, что химические вещества из угля будут конкурентоспособными с нефтехимическими продуктами, когда цена на сырую нефть с низким содержанием серы вырастет вдвое (Richards, 1976).
При такой неопределенности недостаточно веских фактов, чтобы категорически утверждать, что древесина или уголь будут преобладать в качестве альтернативного сырья для химикатов. Конечно, в странах, где есть достаточные запасы древесины и нет угля, или наоборот, выбор будет очевиден.
Заготовка древесины для химической обработки не будет конкурировать с древесиной для пиломатериалов, фанеры и целлюлозы, поскольку она будет зависеть от источников, которые в настоящее время не используются. Ни форма, ни природа древесины для превращения в химикаты не имеют значения.Вид и размер не имеют значения. Например, низкосортная древесина твердых пород на юге Соединенных Штатов, которая не подходит ни для структурных целей, ни для варки целлюлозы из-за размера, вида, дефектов или содержания коры, была бы идеальной для этого применения (Goldstein, 1978). Подобные ресурсы, в настоящее время некоммерческие, имеются в изобилии во всем мире.
Текущая поставленная стоимость такой древесины, заготовленной путем измельчения целых деревьев, может быть намного ниже стоимости щепы балансовой древесины. Однако, если бы он использовался для прямого сжигания для получения энергии, его стоимость могла бы быть ниже, чем стоимость получения того же теплосодержания из альтернативных видов топлива.
ЭНЕРГЕТИКА И ХИМИЯ. Стоимость химикатов, получаемых из сырья, нельзя отделить от энергетических соображений. Например, основными продуктами нефтеперерабатывающих заводов являются бензин и мазут, а не продукты нефтехимии. На цены на нефтехимию влияет внутренний учет, и они были бы намного выше, если бы большая часть накладных расходов не покрывалась основной продукцией.
Аналогичным образом планируется строительство угольных установок для удовлетворения потребности в синтетическом жидком топливе.Химические вещества будут только побочными продуктами. Экономика завода по переработке угля, предназначенного исключительно для химического производства, была бы гораздо: менее благоприятной.
Если обратиться к химическим веществам из древесины, этанол может служить топливом для двигателей внутреннего сгорания, как промышленный химикат сам по себе и как сырье для производства этилена. Рынок для каждого из них разный, но кажется разумным предположить, что цена этанола будет в значительной степени определяться его потенциальным рынком в качестве топлива.
Если побочные продукты варки целлюлозы, такие как лигнин, фенолы и сахариновые кислоты, должны быть извлечены из крафт-черного щелока, они должны быть заменены в целях экономии энергии целлюлозного завода эквивалентным количеством энергии из другого источника.Поскольку одна из функций котла-утилизатора заключается в обеспечении восстановительной атмосферы для неорганических химикатов варки целлюлозы, может потребоваться другой источник углеродных соединений. Водород потребует перепроектирования системы регенерации, а производимого извне технологического пара будет недостаточно, даже если он восполнит дефицит энергии. Это ограничение в сочетании с проблемами разделения налагает серьезные экономические ограничения на большие количества побочных химических продуктов из этого источника.
МАСШТАБ ОПЕРАЦИЙ.В отличие от тысяч миллионов долларов капитальных вложений, требуемых для отдельных нефтехимических заводов мирового масштаба и большой установки по переработке угля, которые считаются необходимыми для экономии на масштабе, для крупной лесохимической промышленности не требуется ни концентрации, ни крупных индивидуальных установок. Необходимость закупки: запас древесины в пределах разумного радиуса вывозки приведет к появлению растений относительно небольшого размера, широко разбросанных по лесным ресурсам. Компромисс между экономией на масштабе и ограничениями на заготовку древесины будет составлять около 2 000 тонн базилика из сухой древесины в день.Это оказалось удобным размером для многих целлюлозных заводов. Предполагаемые капитальные вложения будут в пределах 100 миллионов долларов (Лесная служба Министерства сельского хозяйства США, 1976).
ПРИРОСТ КАПИТАЛА. Поскольку существующие капитальные вложения в нефтехимические предприятия огромны, замена должна быть постепенной, если только внезапная недоступность нефтехимического сырья не потребует срочной программы по их замене. Относительно небольшой размер завода по производству древесных химикатов и соответствующие относительно небольшие требуемые капиталовложения могут повлиять на: решение в пользу использования химических веществ из древесины для увеличения мощности.Это компенсирует неопределенность новых процессов по сравнению с известными нефтехимическими технологиями.
ПОЛИТИЧЕСКИЕ СООБРАЖЕНИЯ. По всей вероятности, решения о строительстве или отказе от строительства завода по переработке древесины в химикаты не будут полностью основываться на технической осуществимости и традиционной экономике предложения, спроса и прибыльности. Государственные стимулы и сдерживающие факторы, несомненно, повлияют на эти решения, особенно с учетом того, что химические вещества и энергия настолько взаимосвязаны.Они могут принимать форму субсидий, поддержки цен, благоприятного налогового режима или налогообложения неблагоприятных альтернативных видов сырья. Социальные решения, принимаемые в разных странах, будут играть важную, а в некоторых случаях и определяющую роль в будущем росте использования химических веществ из древесины.
ЗАЩИТА ОКРУЖАЮЩЕЙ СРЕДЫ. Более широкое использование древесины для преобразования в химические вещества приведет к увеличению лесозаготовительной деятельности, что, в конечном итоге, повлияет на почвы, водосборные бассейны и дикую природу.Поскольку эти же проблемы уже существуют при заготовке древесины для традиционных видов использования, не должно возникнуть особых технических проблем с расширением уже используемых методов.
Однако консервативное отношение общественности к лесной среде может ограничить доступность дополнительной древесины, которая может потребоваться. Состоятельное общество могло бы позволить, чтобы большая часть древесины, которая растет каждый год, перерабатывалась естественными агентами, вместо того, чтобы разрешать урожай этого ежегодного прироста в качестве химического сырья.
ПСИХОЛОГИЧЕСКИЕ БАРЬЕРЫ. Инженеры-химики считают, что с жидкостями и газами работать легче, чем с твердыми веществами, и как нефтяная промышленность, так и сектор химической промышленности, связанный с органическим сырьем, привыкли перекачивать жидкое сырье с помощью танкеров или трубопроводов. Перспектива сбора твердого сырья на тысячи квадратных километров может быть для них настолько чуждой концепцией, что сама ее осуществимость подвергается сомнению.
Кроме того, предприятия лесной промышленности, которые регулярно собирают на своих предприятиях еще большие объемы древесины, в равной степени не согласны с идеей производства химикатов наряду со своими привычными продуктами.
Подобные барьеры исчезнут, когда стимулы станут достаточно большими, но они действительно замедлят процесс конверсии.
После рассмотрения задействованных факторов становится очевидным, что прогнозы относительно будущего химических веществ из древесины могут быть сделаны только в самых общих чертах, и даже тогда они будут сопряжены с неопределенностью. Тем не менее, я осмелюсь высказать свое мнение, хотя оно может быть предвзятым из-за моего личного участия в исследованиях химических веществ из древесины.
Наша нынешняя зависимость от ископаемых углеводородов для производства органических химикатов и полимеров не может продолжаться дольше, чем доступность этого конечного истощаемого ресурса.Даже до полного истощения мировых запасов нефти и газа стоимость этих ресурсов вырастет до уровня, при котором химические вещества из альтернативных ресурсов, таких как уголь и древесина, смогут конкурировать с нефтехимическими продуктами. В одних странах этот момент будет достигнут раньше, чем в других, из-за наличия на местном уровне альтернативных ресурсов и благоприятных политических решений правительства, влияющих на их экономику. Вероятно, что химические вещества как из угля, так и из древесины заменят нефтехимические продукты.
Там, где есть уголь, газификация древесины для производства аммиака, метанола и углеводородов не будет практиковаться, но в отсутствие угля эта технология может быть эффективной. Гидролиз древесины до сахаров для дальнейшего превращения в химические вещества, такие как этанол, фурфурол, молочная кислота и получаемые из них полимеризуемые мономеры, будет преобладающим. Даже сейчас создание интегрированного завода по производству этанола, фенола и фурфурола из твердых пород древесины было бы экономически целесообразным при текущих ценах.
Хотя не будет однозначного ответа на проблему поставок органических химикатов, которая будет вызвана ростом цен на нефть, я считаю, что древесина внесет значительный вклад в ее решение.
ALLAN, G.G. 1978 Производные полимерной целлюлозы. Специальный доклад 8-го Всемирного лесного конгресса.
APPELL, H.R.1971 et al . Преобразование органических отходов в нефть: пополняемый источник энергии. Отчет о расследовании Минного управления 7560 . Департамент внутренних дел США. Вашингтон.
BARTON, G.M. 1978 Химические вещества из деревьев — взгляд в будущее. Специальный доклад 8-го Всемирного лесного конгресса.
БЕНИГНИ, J.D. & I.С. ГОЛЬДШТЕЙН. 1971 г. Нейтральный гидролиз щелочного лигнина до мономерных фенолов. J. Polymer Science , C36, p. 467-475.
Браунинг, Б.Л., изд. 1963 г. Химия древесины. Interscience, 687 с. Нью-Йорк.
CALVIN, M. 1878 Зеленые фабрики. Новости химии и машиностроения , Vol. 56, No. 12, 20 марта, с. 30-36.
ДЭВИС, Д.С. 1978 Меняющийся характер промышленной химии. Новости химии и машиностроения , Vol. 56, № 10, 6 марта, с.22-27.
ГОХИН, Д.В. 1971 г. Низкомолекулярные химические вещества. В: Лигнины, К.В. Сарканен и Ч. Людвиг, ред. Джон Вили, стр. 797-831. Нью-Йорк.
ГОХИН, Д.В. 1972 год. Silvichemicals — какое будущее? Симпозиум Американского института инженеров-химиков 69, стр. 20-24.
GOLDSTEIN, I.S. 1975a Возможности переработки древесины в пластмассу. Наука, Том . 189, стр. 847-852.
GOLDSTEIN, I.S. 1975b Перспективы производства фенолов и фенольных кислот из лигнина и коры. Симпозиум по прикладному полимеру , № 28, с. 259-267.
GOLDSTEIN, I.S. 1976a Химические вещества из лигноцеллюлозы. Biotechnol. и Bioeng. Symp ., № 6, с. 293-301.
GOLDSTEIN, I.S. 1976b Древесина как источник химического сырья. 69-е ежегодное собрание, Американский институт инженеров-химиков, 5 декабря. Чикаго, III.
GOLDSTEIN, I.S. 1977 Место целлюлозы в условиях дефицита энергии. Химия и технология целлюлозы , J.С. Артур, изд. Серия симпозиумов Американского химического общества № 48, стр. 382-387. Вашингтон.
GOLDSTEIN, I.S., D.L. ХОЛЛИ и Э. ИМЕТЬ ДЕЛО. 1978 Экономические аспекты использования низкосортной древесины лиственных пород. Для. Prod. J ., 28. В печати.
HARRIS, J.F. 1975 Реакции кислотного гидролиза и дегидратации для использования растительных углеводов. Симпозиум по прикладному полимеру , № 28, с. 131-144.
HILLIS, W.E. 1978 Экстрактивные вещества . Специальный доклад 8-го Всемирного лесного конгресса.
KATZEN, R.1978 Аспекты исследования химических веществ из древесины: перспективы на будущее. Специальный доклад 8-го Всемирного лесного конгресса.
KUSAMA, J. 1960 Осахаривание древесины с помощью процесса газообразного хлористого водорода. Годовой отчет Ногучинского научно-исследовательского института , 9, стр. 11-22.
LAVER, M.L. 1978 Химикаты из коры. Специальный доклад 8-го Всемирного лесного конгресса.
ЛИПИНСКИЙ, Е.С. 1978 Топливо из биомассы: интеграция с системами питания и материалов.Наука, Vol. 199, стр. 644651.
НАЦИОНАЛЬНЫЙ НАУЧНЫЙ СОВЕТ. 1976 Возобновляемые ресурсы для промышленных материалов. CORRIM Report, Вашингтон, округ Колумбия, Национальная академия наук, 267 стр.
PRAHACS, S., H.G. BARCLAY & S.P. BHATIA. 1971 г. Производство синтетических тоннажных химикатов из остатков лигноцеллюлозы. Pulp Pap. Mag. Канада , 72 (6), стр. 7199-7213.
Робертс Д. 1973 Выращивание светлого дерева у сосен с помощью обработки паракватом. Записка об исследовании лесной службы Министерства сельского хозяйства США SE-191, апрель.Эшвилл, Северная Каролина
САКАНЕН, К.В. 1976 Возобновляемые ресурсы для производства топлива и химикатов, Science, Vol. 191, стр. 773-776.
SCHWEERS, W. 1978 Использование лигнинов, выделенных в мягких условиях из древесины или древесных отходов, для производства полезных химикатов и других химических продуктов. Специальный доклад 8-го Всемирного лесного конгресса.
SEELY, D.B. 1976 Осахаривание целлюлозы для применения в ферментационной промышленности. Biotechnol.и Bioeng. Symp., № 6, с. 285-292.
SHAFIZADEH, F.1978 Разработка пиролиза как нового метода для удовлетворения растущего спроса на продукты питания, химикаты и топливо . Специальный доклад 8-го Всемирного лесного конгресса.
СОЛТЕС, Э.Дж. 1978 Пути термического разложения древесины химическими веществами. Специальный доклад 8-го Всемирного лесного конгресса.
STAMM, A.J. & E.E. HARRIS. 1953 г. Химическая обработка древесины. Chemical Publishing Co., стр. 392-577. Нью-Йорк.
УПРИЧАРД, Дж. М. 1978 Скипидар и талловое масло из лучевой сосны: ценные побочные продукты при варке крафт-целлюлозы. Специальный доклад 8-го Всемирного лесного конгресса.
ЛЕСНАЯ СЛУЖБА USDA. 1976 Возможность использования лесных отходов для производства энергии и химикатов. PB 258 630, Национальная служба технической информации, 193 стр. Спрингфилд, штат Вирджиния.
VAN ANTWERPEN, F.J., 1977 ed. Proc. Конференция по альтернативам химического сырья. Хьюстон, Техас, 2-5 октября 1977 г. Американский институт инженеров-химиков, 125 стр.Нью-Йорк.
WENDER, I., F.W. STEFFGBN & P.W. ЯВОРСКИЙ. 1974 Очистка жидкого и газообразного топлива от твердых органических отходов. В Переработка и удаление твердых отходов , T.F. Йен, шт., Ann Arbor Science Publishers, Inc., стр. 43-99. Анна. Беседка, Ми.
ЦИНКЕЛЬ, Д.Ф. 1975 Военно-морские запасы: лесохимия из сосны. Симпозиум по прикладному полимеру , № 28, с. 309-327.
Оптимальная методология гидролиза бумаги для производства биоэтанола — Журнал молодых ученых
АннотацияВ мире существует большой спрос на топливо, в частности, на экологически чистые виды топлива, такие как биоэтанол.Растет интерес к использованию бумаги в качестве сырья для производства биоэтанола, поскольку это дает двойной эффект: удаление отходов со свалок и удовлетворение глобальных потребностей в энергии. Бумага подвергается кислотному или ферментативному гидролизу, при котором полимеры целлюлозы расщепляются на сахара и ферментируются до этанола. Целью этого исследования было оценить оптимизацию обоих методов гидролиза путем сравнения выходов производства этанола. Стандартная чистая бумага для принтера была измельчена и подвергнута гидролизу с использованием кислотного или ферментативного гидролиза.Затем образцы ферментировали до тех пор, пока реакция не была визуально определена как завершенная в инкубаторе. Для определения количества полученного этанола измеряли плотность и температуру кипения образцов. Не было образцов, которые использовали кислотный гидролиз, который давал бы какой-либо этанол, в то время как один из образцов ферментативного гидролиза произвел приблизительно 28,16 мл этанола. Полученные данные показали, что не было значительной разницы в количестве этанола, полученного в результате кислотного или ферментативного гидролиза бумажного сырья.
ВведениеВ 2018 году Соединенные Штаты потребили 142,86 миллиарда галлонов готового автомобильного бензина, из которых 14,38 миллиарда галлонов пришлось на этанол, который является основным компонентом большинства типов бензина. [1] Действительно, 98% всего бензина в США содержит этанол, и по мере роста мирового населения будет расти и спрос на топливо, эффективность которого аналогична бензину. [2] Концентрации этанола обычно находятся в диапазоне от 10% до 85% (от E10 до E85) и в некоторых случаях до 100%. [1] Этанол — прекрасная альтернатива бензину, поскольку он намного безопаснее для окружающей среды; Фактически, биоэтанол, обычно производимый из кукурузы или сахарного тростника, считается нейтральным по отношению к углероду в атмосфере, поскольку биомасса, из которой производится этанол, поглощает больше CO 2 во время роста, чем произведенный этанол выделяет во время сгорания. [3] Таким образом, этанол на основе биомассы будет экологически устойчивым и теоретически возобновляемым.
Сырье, используемое для превращения в этанол, известно как сырье, наиболее распространенным из которых в настоящее время является кукуруза; 95% этанола в США производится из кукурузы. [2] Однако это занимает значительные площади земли, которые в противном случае можно было бы использовать для выращивания кукурузы в качестве пищи, а не топлива. Еще одним сырьем для производства биоэтанола является бумага, которая привлекательна своей распространенностью и потенциальным использованием отходов. В 2017 году бумага и картон составили 25% (67,01 миллиона тонн) от общего объема 267,8 миллиона тонн твердых бытовых отходов в США. [4] Кроме того, к концу рабочего дня выбрасывается 45% бумаги, печатаемой в офисах. [5] Повторное использование бумаги в качестве сырья для производства биоэтанола может значительно сократить количество отходов. Действительно, биотопливо второго поколения из этой лигноцеллюлозной биомассы получает все большее распространение в промышленности благодаря значительному сокращению выбросов парниковых газов.
Таким образом, использование бумаги в качестве сырья для производства биоэтанола имеет много потенциальных преимуществ. Использование одной тонны переработанной бумаги позволит сэкономить около 682,5 галлона нефти, 26 500 литров воды и 17 деревьев. [6] Этанол можно производить с использованием существующих материалов, устраняя необходимость выращивать кукурузу специально для топливных целей и предоставляя больше земли для сельского хозяйства. Во-вторых, этанол, производимый из бумаги, поможет удовлетворить мировой спрос на бензин, поскольку его можно будет интегрировать в бензин, используемый для двигателей автомобилей и других машин. Тестирование оптимального метода гидролиза бумаги при производстве биоэтанола может помочь компаниям, поскольку они создают для этой цели крупномасштабные предприятия.
Целью данного исследования является поиск оптимального метода гидролиза для производства биоэтанола с использованием бумаги в качестве сырья путем сравнения ферментативного гидролиза с гидролизом серной кислоты (H 2 SO 4 ).Во время гидролиза бумаги целлюлоза расщепляется на сахара в результате реакции с водой. Целлюлоза в бумаге должна быть расщеплена на глюкозу до того, как раствор сможет ферментировать, потому что целлюлоза представляет собой полимер глюкозы, который очень большой и прочный, со связями между мономерами глюкозы, которые не могут быть разрушены и ферментированы дрожжами. Глюкоза, выделяющаяся при гидролизе, должна быть сброжена с использованием дрожжей для производства этанола и диоксида углерода. Затем дистилляция очищает этанол путем кипячения ферментированного раствора при температуре от 100 ℃ (212 ℉) до 78 ℃ (173), чтобы вызвать испарение и специфическую конденсацию в трубчатом аппарате для сбора конечного продукта.Здесь предполагается, что кислотный гидролиз приведет к более высокому выходу, чем ферментативный гидролиз, поскольку кислотный гидролиз в настоящее время является стандартом исследований при использовании бумажного сырья. Кроме того, существует несколько способов проведения ферментативного гидролиза, которые могут повлиять на результаты между различными исследованиями и затруднить обобщение того, какой ферментативный гидролиз является трудным. Например, ферментативный гидролиз можно проводить с использованием нескольких типов ферментов или раствора ферментного коктейля. [7] [8] Если в разных исследованиях используются разные ферменты и разные смеси ферментов, будет трудно узнать, какой фермент является стандартом при использовании ферментативного гидролиза, и вызваны ли различия в данных в разных исследованиях разными ферментами или различиями. в процедурах или других переменных.Потребность в этих исследованиях возрастает по мере роста глобального спроса на топливо и более экологически чистое топливо.
Обзор литературыHtway, Thein и Hlaning сосредоточились на производстве биоэтанола из офисной макулатуры с использованием кислотного гидролиза и ферментации. Были использованы различные количества макулатуры, серной кислоты и дрожжей, чтобы найти оптимальные условия для максимального увеличения выхода этанола.
Кислотный гидролиз использовался как более быстрая и менее дорогая альтернатива ферментам.Сырье было доставлено из офисов и копировальных аппаратов и разрезано на более мелкие части, что привело к большему соотношению площадь поверхности: объем для увеличения скорости реакции. В каждом эксперименте использовали 5 г, 10 г или 15 г нарезанной макулатуры. Бумагу смешивали либо с 100 мл, либо с 200 мл серной кислоты в колбах Эрленмейера на 250 мл. Гидролиз проходил в автоклаве при 121 ° C.
Полученный раствор фильтровали для удаления твердых частиц и добавляли гидроксид натрия для поддержания оптимума pH 4.5-5 для ферментационной активности дрожжей Saccharomyces cerevisiae . S. cerevisiae использовали в различных количествах при 30 ° C в течение 24 часов. Оптимальные условия для производства биоэтанола: 10 г офисной макулатуры, гидролиз через 100 мл 5% H 2 SO 4 при 121 ° C в течение 60 минут и 2 г дрожжей.
Как и в большинстве исследований по этой теме, в этом исследовании использовалась слабая кислота для процесса гидролиза, превращающего целлюлозу в глюкозу. Однако перегонка их продукта не проводилась, иначе она могла бы очистить и облегчить дальнейшие испытания биоэтанола.Еще одно ограничение заключалось в отсутствии определенного процесса нейтрализации или предварительной обработки.
Чтобы рассмотреть отдельные этапы процесса производства этанола, исследования гидролиза бумаги были оценены Масейрасом, Альфонсином и Пулом. Подобно Htway, Thein и Hlaing использовали кислотный гидролиз. Макулатуру канцелярской бумаги собирали и разрезали на куски площадью 0,25 см 2 . 5 г подготовленной бумаги помещали в колбу с различными объемами 5% H 2 SO 4 .Использовали 100 мл, 200 мл или 300 мл кислоты, и растворы помещали в автоклав при 121 ° C. В отличие от большинства исследований, время варьировалось в дополнение к количеству кислоты, и время гидролиза составляло 30 минут, 60 минут, 120 минут или 180 минут. Затем раствор фильтровали, центрифугировали и раствор нейтрализовали до pH от 4,5 до 5, используя 5М раствор гидроксида натрия.
Ферментация происходила при 30 ° C в течение 24 часов при 150 об / мин орбитального встряхивания смеси гидролизованного раствора, 15% ферментационной среды и 10% дрожжевой суспензии.Затем ферментированный раствор дважды перегоняли для получения биоэтанола оптимального качества. Затем исследователи протестировали биоэтанол на предмет определенных свойств, таких как плотность, объем кислоты, цвет, мутность и показатель преломления, чтобы охарактеризовать биоэтанол. Они пришли к выводу, что идеальное количество кислоты для гидролиза составляет 200 мл, в то время как идеальное время для гидролиза составляет 120 минут.
Несмотря на то, что было шесть разных образцов, только один из них имел 100 мл H 2 SO 4 и только один образец был 300 мл H 2 SO 4 .Исследователи должны были иметь несколько образцов для каждого количества H 2 SO 4 для более стабильных результатов. Это исследование было похоже на многие другие исследования по этой теме с использованием кислотного гидролиза вместо ферментативного гидролиза и их процесса, который включал предварительную обработку, гидролиз, нейтрализационную ферментацию и дистилляцию. Одно отличие от этого исследования заключалось в том, что оно изменяло количество времени для гидролиза, тогда как во многих исследованиях изменялось только количество кислоты или ферментов, используемых в процессе гидролиза.Результаты этого исследования могут быть использованы для более детального изучения других переменных, влияющих на производство этанола из макулатуры. Наше исследование будет сосредоточено на различии между ферментативным гидролизом и кислотным гидролизом при производстве этанола. В отличие от этого исследования, наше исследование не будет использовать отклонения во времени на стадии гидролиза. Однако, как и в этом исследовании, в нашем исследовании будут использоваться гидролиз, нейтрализация, ферментация и дистилляция.
Wang et al. в 2013 году провела исследование ферментативного гидролиза макулатуры при производстве биоэтанола.В качестве сырья в исследовании использовались газеты, офисная бумага, журналы и картон. Cellic Ctec 1 представлял собой ферментный комплекс, используемый для гидролиза. Макулатуру смешивали с водой в концентрации 15% (мас. / Мас.) В течение 10 минут. Серную кислоту добавляли для поддержания pH раствора на уровне 5,2, и процесс осахаривания занял 72 часа при 50 ° C. На протяжении всего процесса конверсии измеряли и сравнивали выходы различных типов сахара (глюкозы, ксилозы, целлобиозы, галактозы, маннозы и арабинозы) для разных типов бумаги.
Бумажное сырье было разделено на две категории: базовые сценарии и современные сценарии. Базовый вариант использовался для макулатуры; приготовлены путем измельчения, ферментативного гидролиза при 50 ° C в течение 72 часов, ферментации при 40 ° C в течение 36 часов с использованием бактерий Zymomonas mobilis с диаммонийфосфатом (DAP), кукурузным настоем (CSL) и оборотной водой и, наконец, дистиллированной для получения 99,5% (мас. / мас.) биоэтанола. Диаммонийфосфат (DAP) и кукурузный настой (CSL) добавляли в соответствии со специальным процессом ферментации, установленным Национальной лабораторией возобновляемых источников энергии (NREL).
Современный футляр использовался для офисной бумаги и газет. Офисную бумагу измельчали и нагревали до 100 ° C паром низкого давления, после чего добавляли 5% (мас. / Мас.) H 2 SO 4 при 220 ° C в течение 2 минут. H 2 SO 4 добавляли к раствору, чтобы довести pH до 5, раствор фильтровали, детоксифицировали путем удаления льда и затем снова фильтровали. Затем раствор гидролизовали жидкими ферментами Cellic Ctec 1 и ферментировали диаммонийфосфатом (DAP), кукурузным настоем (CSL), оборотной водой и Zymomonas mobilis .Затем ферментированный продукт перегоняли с получением биоэтанола. В случае с современной газетой последовал более сложный набор шагов по сравнению с делом о современной офисной бумаге. Газету измельчали и нагревали до 100 ° C паром низкого давления, затем гидролизовали гидроксидом кальция (CaOH 2 ), нейтрализовали и фильтровали. Затем раствор прошел тот же процесс гидролиза, ферментации и дистилляции, что и современный футляр для офисной бумаги, для получения биоэтанола.
Затем исследователи провели экономический анализ, рассчитав общую стоимость проекта, операционные расходы и дисконтированный денежный поток. Результаты показали, что офисная бумага имеет самый высокий выход глюкозы, за ней следуют картон, газеты и журналы. Кроме того, большая часть затрат на эксперименты приходилась на биомассу, и лишь небольшая часть затрат приходилась на ферменты. Сравнивая биоэтанол и бензиновый бензин, исследователи обнаружили, что все, кроме биоэтанола, произведенного из журнальной бумаги, было экономически лучше, чем цены на бензиновые насосы.
Исследование было очень тщательным в их экспериментировании, добавляя многочисленные этапы и процессы в производстве биоэтанола. Однако достоверность выводов не может быть абсолютной, поскольку разные протоколы, применяемые к разным типам бумаги, не обеспечивают нормализованное сравнение.
Это исследование отличалось от многих других, поскольку исследователи провели обширный экономический анализ процесса производства этанола и биоэтанола, а также варьировали типы используемого бумажного сырья.В исследовании также использовались дрожжи и ферменты другого типа, чем в большинстве исследований. Большинство исследований также измеряли только выход глюкозы, а не выход любых других типов сахара в процессе гидролиза.
В отличие от других исследований, Kang et al. вела производство биоэтанола с использованием смеси сосновой щепы и макулатуры в качестве сырья, полученного из промышленных побочных продуктов компании Rock-Tenn и бумажной фабрики Kraft соответственно. Сырье было проанализировано на содержание влаги, углеводов и золы.Цель заключалась в создании метода производства этанола, который был бы одновременно возобновляемым и уменьшающим количество отходов.
Исследователи выполнили ряд шагов, которые очень напоминают шаги, используемые в других исследованиях. Сначала они создали целлюлозу или смесь бумаги, сосновой стружки и воды. Бумажный шлам подвергали ферментативному гидролизу в течение 48 часов при 50 градусах Цельсия, чтобы убедиться, что было преобразовано как можно больше целлюлозы. Затем исследователи добавили дрожжевой раствор к бумажному шламу и поместили его в шейкер инкубатора, где он ферментировал при 36 градусах Цельсия, а вращался со скоростью 150 об / мин.Образцы отбирали из шейкера инкубатора с 6, 12, 24 и 48-часовыми интервалами и анализировали на содержание сахара и этанола. Вместо ферментации, основанной на определенных временных приращениях, можно использовать колбу для ферментации при комнатной температуре, и образцы могут быть удалены, когда колба указывает на окончание процесса ферментации. Исследователи никогда не очищали этанол, предпочитая измерять содержание этанола в растворе после ферментации. Дистилляция может быть использована в будущих исследованиях для создания более чистого биоэтанола.
В этой статье отсутствуют некоторые очень важные шаги, которые также следует выполнять, такие как дистилляция и использование фермента целлюлазы. Также было представлено много новых способов проведения шагов. Например, тот факт, что существует оптимальная температура брожения, может быть учтен в нашем исследовании.
Бяджи и Калбурги представили способ производства биоэтанола из газетных отходов с использованием смеси серной кислоты и газет в соотношении 10: 1. Затем исследователи использовали дрожжи, чтобы сбродить раствор в течение 36 часов, создав этанол.Наконец, они отогнали этанол, чтобы создать чистый образец. Этот метод очень похож на метод, который используется в других исследованиях, и применим к нам, хотя некоторые шаги можно было бы удалить, а другие добавить.
Во-первых, исследователи собрали отходы газет из домашних хозяйств, они удостоверились, что они не подвергались воздействию большого количества пыли или грибков, были высушены на солнечном свете и хранились в запечатанных пластиковых пакетах, чтобы предотвратить любые искажающие переменные. Затем они взяли бумагу, измельчили ее на «маленькие» кусочки и поместили их в запечатанный пакет перед предварительной обработкой.Предварительная обработка образцов началась с того, что исследователи добавили смесь серной кислоты (H 2 SO 4 ) в бумажный водный раствор. Этот шаг был необходим, чтобы разбить бумагу на целлюлозные цепочки для лучшего гидролиза. Гидролиз предварительно обработанного субстрата не был разъяснен в статье, но фермент под названием целлюлаза может быть использован для расщепления целлюлозы на глюкозу. Затем гидролизованный «бульон» смешивали с коммерческими дрожжами, где «бульон» ферментировали при 34 градусах Цельсия в течение 36 часов, создавая очень разбавленный биоэтанол.Вместо того, чтобы процесс ферментации зависел от времени, можно использовать колбу для ферментации, чтобы показать исследователям, когда процесс ферментации закончился. В статье не говорится, как они преобразовали разбавленный биоэтанол в более чистую форму, остановившись после процесса ферментации. Дистилляция может использоваться для отгонки этанола с помощью перегонной трубки, потому что этанол кипит при гораздо более низкой температуре, чем вода, поэтому это был бы приемлемый метод очистки «бульона» биоэтанола.
В этой статье дается подробное объяснение того, как преобразовать газетные отходы в разбавленный биоэтанол, но было бы более полезно, если бы исследователи дистиллировали биоэтанол или каким-либо образом разделяли субстрат и этанол.Однако большинство процессов можно воспроизвести в нашем исследовании.
МетодологияЦелью данного исследования было сравнение количества этанола, полученного каждым методом гидролиза; таким образом, независимая переменная — это протокол для ферментативного и кислотного гидролиза, а зависимая переменная — это выход этанола, измеренный как приблизительный процент и объем. Для получения этанола были протестированы два метода гидролиза бумаги. Ферменты целлюлазы и 6% -ная серная кислота использовались для ферментативного и кислотного гидролиза соответственно.Методология этого исследования была разделена на пять компонентов: предварительная обработка, гидролиз, ферментация, дистилляция, а также тесты и анализ данных. В большинстве рассмотренных выше исследований использовался схожий общий процесс и набор шагов для создания этанола из бумаги.
Перед экспериментальной реализацией сырье было предварительно обработано. Стандартная чистая бумага для принтера использовалась, чтобы избежать использования чернил как мешающей переменной, которая может химически повлиять на процесс гидролиза. Вся бумага была измельчена, а затем разделена на полфунта или 227 г (3 шилл.е.) образцы. Все исследования, перечисленные выше в обзоре литературы, либо разрезали бумагу на более мелкие кусочки, либо смешивали ее в массу мелких кусочков бумаги. Масса бумаги, используемой для кислотного и ферментативного гидролиза, различалась из-за насыщения субстрата для каждого конкретного реагента.
Один образец кислотного гидролиза подвергали гидролизу при комнатной температуре, но все другие образцы кислотного гидролиза проводили в скороварке. Для кислотного гидролиза к бумажному сырью по весу добавляли 6% -ную серную кислоту в соотношении 5: 1 или 10: 1.6% серной кислоты использовалось для того, чтобы внимательно следить за всеми изученными исследованиями, в которых использовался кислотный гидролиз, в котором использовалось 5%. Отношение кислоты к бумаге также использовалось по аналогии с обзорными исследованиями, в которых Maceiras et al. использовали соотношение 4: 1, и оба Htway et al. и Byadgi et al. используется соотношение 10: 1. В скороварке поддерживалась температура около 100 ℃ (212 ℉), и она работала в течение примерно одного часа каждый день с 10-фунтовым пароотводным клапаном для создания давления в контейнере. Полный гидролиз длился между 47.5 и 360 часов. Для отделения оставшейся суспензии от жидкого раствора использовали сетчатый фильтр. Затем раствор перемешивали и нейтрализовали добавлением двух концентраций гидроксида натрия: более сильной концентрации в форме таблеток и более низкой концентрации в жидкой форме. PH контролировали с помощью цифрового pH-метра и добавляли гидроксид натрия до тех пор, пока pH раствора не стал приблизительно нейтральным (pH между 6 и 7), чтобы гарантировать, что образцы были оптимизированы для ферментации, чтобы дрожжи не были слишком кислыми или слишком простая среда, которая составляла от 40 до 43 граммов таблеток и от 0 до 15 мл жидкости.
Ферментативный гидролиз включал добавление 4 г целлюлазы в открытый контейнер с бумажным сырьем и от 3,6 до 6 л воды для предотвращения реакций с ограниченной скоростью. После того, как один образец ферментативного гидролиза подвергся гидролизу под нагревательной лампой, гидролиз позже был проведен в инкубаторе, чтобы обеспечить равномерно нагретую среду ниже 40 ℃ для оптимального функционирования дрожжей, как показано в протоколах из обзора литературы. Переключение с нагревательной лампы на инкубатор произошло потому, что нагревательная лампа нагревает только верхнюю часть гидролизующего раствора, тогда как инкубатор обеспечивает равномерный нагрев всего раствора.Гидролиз длился от 120 до 408 часов, чтобы целлюлаза расщепила целлюлозу на сахар. Поскольку в ферментативном гидролизе не участвовали кислоты, нейтрализовать раствор не требовалось. По окончании ферментативного гидролиза бумагу отделяли от образца и добавляли жидкость в колбу для ферментации.
Процесс ферментации был одинаковым для образцов кислотного и ферментативного гидролиза и проходил в инкубаторе, который был установлен ниже 40 ℃. Ферментацию проводили в инкубаторе, который был установлен ниже 40 ℃ в соответствии с Wang et al., Kang et al. И Byadgi et al. Для брожения в раствор добавляли минимум 5 г стандартных пекарских дрожжей и останавливали его S-образной пробкой с двумя пузырьками (показана на рисунке 1), которую заполняли водой для наблюдения за образованием газа CO 2 в виде подтверждение успешного брожения. Минимальное время для ферментации составляло семь дней, в течение которых химический стакан следует осторожно встряхивать два или три раза в день, чтобы ускорить процесс и обеспечить равномерное распределение дрожжей в растворе.Если визуальные признаки ферментации сохранялись через семь дней, ферментация продолжалась до исчезновения визуальных признаков.
Рис. 1 : Схема S-образной пробки воздушного шлюза с двумя пузырьками.
После завершения ферментации ареометр использовался для проверки наличия этанола перед дистилляцией. Дистилляция также использовалась Масейрасом и др. и Wang et al. Температуру смеси поддерживали между точками кипения чистого этанола и чистой воды (78.4 ℃ (3sf.) и 100 ℃ соответственно), чтобы обеспечить разделение жидкостей. Во время процесса дистилляции этанол (и немного воды) испарялся, поднимался до того места, где он встречался с холодной дистилляционной трубкой, конденсировался обратно в жидкость и направлялся в небольшой мерный цилиндр или небольшой химический стакан для сбора. Посредством серии предэкспериментальных тестов, чтобы лучше понять дистилляцию, было обнаружено, что при испарении этанола жидкость конденсируется в гладкие полосы. В то время как при испарении воды пар конденсировался в отдельные капли, которые плавились и оставались неподвижными до достижения порогового максимального объема, а затем падали в градуированный цилиндр.Обычно, если в образце присутствуют следы этанола, этанол отгоняют в течение первого часа перегонки. Образец отбрасывали, если этанол не отгонялся, что подтверждается перегонкой, происходящей только при температуре около 100 ℃, и тем, что полученная жидкость не горит при воздействии пламени. Когда образец жидкости был собран и показал признаки содержания этанола, его затем подвергли серии перегонок для получения максимального процентного содержания этанола, обычно два-три раза.
Затем конечный продукт собирали и тестировали для измерения точек кипения и плотности, чтобы охарактеризовать долю этанола в конечном продукте. Температура жидкости измерялась с интервалами, поскольку разные температуры могут изменять плотность образца. После этого регистрировали температуру, при которой образец начинал кипеть, чтобы гарантировать, что измерение плотности было более точным из-за сохраняемого объема этанола. После того, как все конечные продукты были собраны и проанализированы, был использован критерий значимости Манна-Уитни для сравнения двух методов гидролиза, чтобы увидеть, какой метод дает больший выход этанола.
РезультатыДля этого исследования было проверено и сравнено количество этанола, полученного с помощью кислотного и ферментативного гидролиза. Были завершены два образца кислотного гидролиза и три образца ферментативного гидролиза. Результаты экспериментов по производству биоэтанола с использованием как кислотного, так и ферментативного гидролиза показаны на рисунке 2. Оба образца после кислотного гидролиза не дали этанола. К такому выводу пришли во время перегонки, когда жидкость не перегонялась из круглодонной колбы при температуре ниже 210 ° С.Любая жидкость, которая вышла наружу, имела температуру кипения примерно при температуре кипения воды (212 ℉ или 100 ℃) и была негорючей. Кроме того, любые жидкие пары, которые конденсировались в дистилляционной трубке, не имели длинного плавного хода, характерного для этанола. Из-за этих факторов не было необходимости проверять плотность или температуру кипения этих образцов, поскольку в этих образцах приблизительно не было этанола. Два из трех образцов ферментативного гидролиза дали те же результаты, что и образцы кислотного гидролиза.Последний образец ферментативного гидролиза действительно произвел конечный продукт для тестирования.
| Кислотный гидролиз (выход этанола в мл) | Ферментативный гидролиз (выход этанола в мл) |
| 0 | 0 |
| 0 | 0 |
| 28,16 * |
Рисунок 2 : Результаты экспериментов по производству биоэтанола. В левом столбце показано количество этанола в миллилитрах от производства биоэтанола с использованием кислотного гидролиза.В правом столбце представлено количество этанола в миллилитрах от производства биоэтанола с использованием ферментативного гидролиза.
Измерена плотность третьего образца ферментативного гидролиза. Температура жидкости при измерении плотности составляла 66,4 (19,1 ℃), масса образца составляла 30 граммов, а объем составлял приблизительно 35,2 мл. Плотность была рассчитана как 0,852. Чтобы получить приблизительное процентное содержание этанола в жидкости, на рисунке 3 использовалась плотность в сочетании с температурой.Используя рисунок 3, было показано, что температура образца была наиболее близка к 20 ℃, а плотность была между 0,843 и 0,868. Тест на плотность утверждает, что образец содержал от 70% до 80% этанола по весу.
| этанол вес (%) | Температура (° C) | ||||||
| 10 | 15 | 20 | 25 | 30 | 35 | 40 | |
| 0 | 1.000 | 0.999 | 0,998 | 0,997 | 0,996 | 0,994 | 0,992 |
| 10 | 0,984 | 0,983 | 0,982 | 0,980 | 0,979 | 0,977 | 0,975 |
| 20 | 0,972 | 0,971 | 0,969 | 0,966 | 0,964 | 0,961 | 0,959 |
| 30 | 0,960 | 0.957 | 0,954 | 0,951 | 0,947 | 0,944 | 0,941 |
| 40 | 0,942 | 0,939 | 0,935 | 0,931 | 0,928 | 0,924 | 0,920 |
| 50 | 0,922 | 0,918 | 0,914 | 0,910 | 0,906 | 0,902 | 0,897 |
| 60 | 0,899 | 0.895 | 0,891 | 0,887 | 0,883 | 0,878 | 0,874 |
| 70 | 0,876 | 0,872 | 0,868 | 0,863 | 0,859 | 0,855 | 0,850 |
| 80 | 0,852 | 0,848 | 0,843 | 0,839 | 0,835 | 0,830 | 0,826 |
| 90 | 0,827 | 0.822 | 0,818 | 0,814 | 0,809 | 0,805 | 0,800 |
| 100 | 0,798 | 0,794 | 0,789 | 0,785 | 0,781 | 0,776 | 0,772 |
Рисунок 3 : Плотность смесей этанола и воды из образцов, полученных в результате ферментативного гидролиза. Крайний левый столбец представляет собой процентное содержание этанола. Верхний ряд представляет температуру смеси в градусах Цельсия.
28,16 — это приближение, основанное на тестах на плотность и температуру кипения. Тест на плотность показал, что образец содержал от 70% до 80% этанола. Тест на точку кипения показал, что образец содержал от 80% до 90% этанола. Взяв среднее значение из двух прогнозируемых процентов из двух тестов, мы утверждаем, что образец примерно на 80% состоит из этанола. Поскольку объем образца составлял приблизительно 35,2 мл, 80% от 35,2 составляет 28,16.
Также был проведен тест на точку кипения.Красная точка на рисунке 4 представляет точку кипения третьего образца ферментативного гидролиза. Используя рисунок 4, было показано, что образец содержал от 80% до 90% этанола по объему на основе теста точки кипения.
Рис. 4 : Температура кипения смесей этанола и воды. Нижняя ось абсцисс представляет процентное содержание этанола в жидкости. По оси ординат отложена температура кипения жидкости в градусах Цельсия и Фаренгейта.
После того, как все образцы были собраны, U-критерий Манна-Уитни был использован для оценки гипотезы этого исследования.Этот тест использовался из-за небольшого размера выборки (<5), что означало, что нельзя было предполагать нормальность популяции. Нулевая гипотеза заключалась в том, что не было разницы в выходах этанола между производством биоэтанола в результате кислотного гидролиза и ферментативного гидролиза. Альтернативная гипотеза заключалась в том, что при производстве биоэтанола с использованием кислотного гидролиза производится больше этанола, чем с использованием ферментативного гидролиза. Использовался правосторонний тест со значением альфа 0,05. При использовании данных этого исследования в тесте значимости U Манна-Уитни действуйте с осторожностью, потому что размеры выборки для двух методов гидролиза были меньше 5.Результаты теста значимости показаны на рисунке 5. Поскольку значение p было больше, нулевая гипотеза была принята; было достаточно доказательств, чтобы предположить, что не было значительной разницы в выходе этанола при кислотном и ферментативном гидролизе.
| Данные критерия значимости U Манна-Уитни |
Результат 0,889664> 0,05, не отклонить |
Рис. 5 : Данные для критерия значимости U Манна-Уитни и результаты критерия значимости.
ОбсуждениеОграниченный размер выборки может быть связан с трудностями в поддержании определенных условий; на ранних этапах тестирования токсичность этанола возрастала во время ферментации до тех пор, пока дрожжи не умерли и процесс ферментации не прекратился, тем самым ограничивая количество сахара, превращаемого в этанол.Кроме того, был только один образец с прослеживаемым содержанием этанола, что потенциально могло представлять ошибку в процедуре.
Это исследование применимо только для оптимального производства биоэтанола из бумаги с использованием ферментативного или кислотного гидролиза. Это исследование не применимо к оптимальному количеству ферментов или кислоты, необходимых для гидролиза, времени ферментации, времени дистилляции, дрожжей для ферментации, соотношению бумаги и кислоты или бумаги к ферменту, или оптимальной температуре во время гидролиза или ферментации.
Это исследование может быть расширено путем определения таких параметров, как оптимальная масса ферментов или объем кислоты, необходимый для гидролиза, экспериментальные условия, виды используемых дрожжей или соотношение сырье: реагент.
Основным ограничением этого исследования было время. В начале этого исследования на сбор данных предполагалось четыре с половиной месяца, которые закончились преждевременно из-за пандемии COVID-19. Исключение этих образцов потенциально исказило результаты этого исследования.Кроме того, количество и тип оборудования были ограничены; Некоторое оборудование, используемое в подобных исследованиях, такое как автоклавы, среды для ферментов целлюлазы и рефрактометры, не было доступно для использования в этом исследовании.
Ареометр создал путаницу в исследовании, которую можно было бы лучше оценить в будущих исследованиях. Каждый раз, когда ареометр использовался после завершения ферментации образца, он показывал, что в образце не было этанола. Однако, когда ареометр был протестирован на других небольших боковых испытаниях, он работал правильно.Даже при отрицательных результатах ареометра образцы все равно подвергались перегонке. Ложноотрицательные результаты ареометра сбивали с толку, когда ферментация была фактически завершена и действительно ли в образце присутствовал этанол.
Для улучшения процедуры этого исследования было бы полезно рассчитать содержание сахара после гидролиза для обоих методов. Расчет содержания сахара после гидролиза покажет, какой метод дает больше сахара и, следовательно, теоретически больше этанола.Затем методы гидролиза можно было сравнить, чтобы определить, какие из них производили больше сахара во время гидролиза, а какие — больше этанола. Теоретически метод гидролиза, при котором образуется больше всего сахара, должен производить больше всего этанола. Если этого не произойдет, это может быть возможностью исследовать другие факторы, которые могут повлиять на количество этанола, производимого после стадии гидролиза. Есть два способа измерить содержание сахара в растворе; рефрактометр может быть использован для измерения ° Bx для представления содержания сахарозы, или восстанавливающие сахара могут быть измерены с использованием динитросалициловой кислоты как часть метода DNS. [9] [10]
Для будущих исследований рекомендуется использовать небольшое количество бумаги для каждого образца. Во время этого исследования было быстро замечено, что использование бумаги размером в полфунта было слишком большим, что делало процесс дорогостоящим и затрудняло поддержание в правильном аппарате для каждой реакции. Также может быть полезен мониторинг pH образцов во время ферментации, поскольку раствор становится более кислым, убивая дрожжи при pH ниже 4. Добавление большего количества дрожжей в ферментирующий раствор не приведет к повторной стимуляции ферментации из-за токсичности раствора после определенного порог этанола был создан.Таким образом, для поддержания активности дрожжей в раствор следует добавить буфер, такой как бикарбонат натрия или бикарбонат калия. Другой вопрос, который можно было бы проверить в будущих исследованиях, — какой метод гидролиза является наиболее простым и наименее дорогостоящим. Ферментативный гидролиз казался намного проще, потому что не было необходимости гидролизовать в автоклаве или нейтрализовать раствор после гидролиза. Это может быть значительным в промышленных приложениях, где на крупномасштабных предприятиях требуются дополнительные этапы и затраты на автоклавы и нейтрализующие химикаты с кислотным гидролизом.
ЗаключениеИз проверки гипотезы можно сделать вывод, что нет достаточных доказательств того, что при производстве биоэтанола с использованием кислотного гидролиза образуется больше этанола, чем с использованием ферментативного гидролиза. Первоначальная гипотеза о том, что при производстве биоэтанола с использованием кислотного гидролиза дает больше этанола, не была доказана, но и не была опровергнута; предыдущие исследователи продемонстрировали высокие выходы с помощью методов кислотного гидролиза, но это не нашло отражения в данном исследовании.Однако из-за ограниченного размера выборки достоверность этих результатов должна быть дополнительно подтверждена увеличением размера выборки в более поздних экспериментах. Достоверность этих результатов также может быть ограничена из-за различных концентраций исходного сырья, так как сравнение результатов по разным образцам может стать затруднительным. Результаты этого исследования нельзя сравнивать с другими исследованиями, потому что нет известных исследований, сравнивающих производство биоэтанола с использованием кислотного и ферментативного гидролиза. Потребуется провести дополнительные исследования, чтобы подтвердить или опровергнуть результаты этого исследования.Хотя результаты этого исследования не предоставили убедительных доказательств того, какой метод гидролиза дает больше этанола, он все же может быть применим и ценен для будущих исследователей, которые хотят сравнить методы гидролиза для производства биоэтанола с бумагой. Поскольку мир ищет новые, экологически безопасные способы удовлетворения своих потребностей в топливе с использованием материалов многократного использования, поиск наиболее эффективного и действенного метода гидролиза окажется неоценимым для крупномасштабного производства биоэтанола с бумагой в качестве сырья.
БлагодарностиОсобая благодарность доктору Курту Гутеру за то, что он потратил свое время на помощь в этом исследовании своим опытом. Особая благодарность г-ну Джо Расмусу за приобретение необходимых материалов для этого исследования и советы.
ПриложениеПолный список всех материалов, использованных в этом исследовании, приведен ниже:
- Уничтожитель документов
- Гранулы гидроксида натрия и жидкость для нейтрализации после гидролиза
- Горелка Бунзена для перегонки.
- Круглодонная колба на 500 мл
- Круглая колба для кипячения 150 мл
- Круглая колба на 1000 мл с двумя горловинами для перегонки
- Цифровой термометр для измерения температуры жидкого раствора во время перегонки и испытаний точки кипения
- Дистилляционная колонна 500 мм
- Шлюз и пробка с двумя пузырьками S-образной формы
- Весы для взвешивания бумаги, ферментов целлюлазы и дрожжей
- Стаканы и градуированные цилиндры различных размеров для измерения воды, серной кислоты, гидроксида натрия и сбора этанола во время дистилляции
- Стаканы 2000 мл для брожения
- Магнитная мешалка и различные магнитные мешалки
- Цифровой pH-метр
- Резиновые перчатки для защиты
- Защитные очки для обеспечения безопасности
- Ферменты целлюлазы (порошок)
- Серная кислота (6%)
- Стандартная чистая бумага для принтера
- Электрическая скороварка для кислотного гидролиза
- Водопроводная вода, используемая при ферментативном гидролизе
- Стандартные активные сухие пекарские дрожжи
- Ареометр 0-100%
- Большой инкубатор
- Стеклянные палочки для перемешивания во время гидролиза
- Большие полиэтиленовые пластиковые открытые емкости для ферментативного и кислотного гидролиза
- Стеклянная емкость на 5000 мл для кислотного гидролиза в скороварке
- Фильтр для удаления остатков бумажного шлама после гидролиза
- «Часто задаваемые вопросы (Faqs) — U.S. Управление энергетической информации (EIA) ». 2020. Eia.Gov . По состоянию на 6 мая. Https://www.eia.gov/tools/faqs/faq.php?id=27&t=10 .
- DOE, США. «Центр данных по альтернативным видам топлива: основы этанольного топлива». Центр данных по альтернативным видам топлива (2012 г.): https://afdc.energy.gov/fuels/ethanol_fuel_basics.html
- «Этанол и окружающая среда — Управление энергетической информации США (EIA)». 2019. Eia.Gov . https://www.eia.gov/energyexplained/biofuels/ethanol-and-the-environment.php
- Агентство по охране окружающей среды США (EPA). «Национальный обзор: факты и цифры о материалах, отходах и вторичной переработке». (2019): https://www.epa.gov/facts-and-figures-about-materials-waste-and-recycling/national-overview-facts-and-figures-materials .
- О’Мара, Морган. 2020. «Сколько бумаги используется за один день». Recordnations.Com . https://www.recordnations.com/2016/02/how-much-paper-is-used-in-one-day/
- «Мир имеет значение». 2019. Theworldcounts.Com . По состоянию на 22 октября. Https://www.theworldcounts.com/stories/Paper-Waste-Facts .
- Кан, Ли, Юн Ю. Ли, Сон Хун Юн, Аллен Дж. Смит и Гопал А. Кришнагопалан. «Производство этанола из смеси предварительного гидролизата гемицеллюлозы и бумажного шлама». Биоресурсы 7, вып. 3 (2012): 3607-3626
- Ван, Лей, Махди Шарифзаде, Ричард Темплер и Ричард Дж. Мерфи. «Производство биоэтанола из различных макулатур: экономическая целесообразность и анализ чувствительности». Приложенная энергия 111 (2013): 1172-1182
- Htway, Wai Mar, Thu Zar Thein и Y.Мят. «Производство биоэтанола из офисной макулатуры». Int. J. Sci. Англ. Заявление 7, вып. 08 (2018): 166-168
- Byadgi, Shruti A., and P. B. Kalburgi. «Производство биоэтанола из газетных отходов». Процедура Науки об окружающей среде 35 (2016): 555-562
- Масейрас Р., В. Альфонсин и Дж. Э. Пул. «Производство биоэтанола из макулатуры офисной бумаги». Международный научный журнал Journal of Environmental Science. 6 (2017): 48-51
- МакФарланд, Томас В. и Ян М. Йейтс.«Ю-тест Манна – Уитни». Во Введение в непараметрическую статистику для биологических наук с использованием R, стр. 103-132. Спрингер, Чам, 2016
- S-образная пробка воздушного шлюза с двумя пузырями https://i5.walmartimages.com/asr/74d5dbf2-8de2-4d38-a137-43fe94ec2bbd_1.312ff377ea6c8f76be128ca128ce6f2e.jpeg?odnWidth=612&odnHeight=Beff
Мэтью Уилсон
Мэтью Уилсону 18 лет, он учится в старшей школе Уильямстона в Уильямстоне, штат Мичиган.Он является членом Академии математики и наук (MSA), которая представляет собой продвинутую программу по математике и естественным наукам для старшеклассников. Он является членом кросс-кантри и легкоатлетических команд. В свободное время Мэтью любит путешествовать с рюкзаком и посещать национальные парки.
Джек Шафер
Джеку Шаферу 17 лет, он учится в Уильямстонской средней школе. Джек является членом Академии математики и наук Уильямстона, где учащимся предоставляются такие возможности, как продвинутые классы математики и естествознания, а также возможность посещать исследовательские классы в младших классах.